




| Short Communication | ||
Open Vet. J.. 2026; 16(1): 711-720 Open Veterinary Journal, (2026), Vol. 16(1): 711-720 Short Communication Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, ChinaNuo Xu1#, Mingrui Ma1#, Hannuo Li1, Zhuolei Yang1, Binglun Sui1, Shaojie Song1, Wanli Sha1,2,3, Baishuang Yin1,2,4, and Wenlong Dong1,2,3*1College of Animal Science and Technology, Jilin Agricultural Science and Technology College, Jilin City, China 2Jilin Provincial Key Laboratory of Preventive Veterinary Medicine, Jilin City, China 3Jilin Province Technology Innovation Center of Pig Ecological Breeding and Disease Prevention and Control, Jilin City, China 4Jilin Province Cross Regional Cooperation Technology Innovation Center of Porcine Main Disease Prevention and Control, Jilin City, China *Corresponding Author: Wenlong Dong. College of Animal Science and Technology, Jilin Agricultural Science and Technology College, Jilin City, China. Email: dongwenlong888 [at] jlnku.edu.cn Submitted: 09/10/2025 Revised: 12/12/2025 Accepted: 27/12/2025 Published: 31/01/2026 © 2025 Open Veterinary Journal
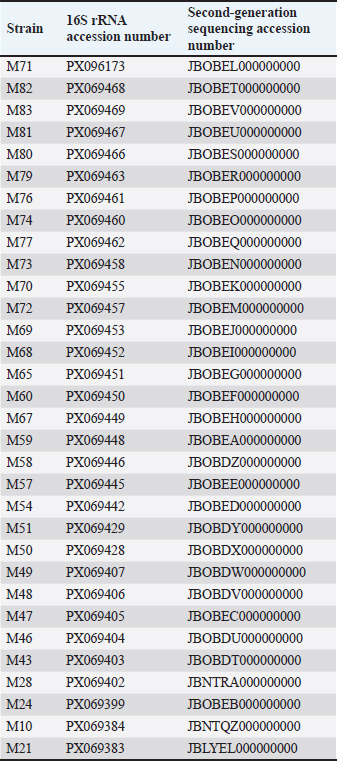
AbstractBackground: Coagulase-negative staphylococci, such as Staphylococcus cohnii are increasingly recognized as crucial reservoirs of antimicrobial resistance (AMR) genes. However, their role in the dissemination of multidrug resistance within livestock populations, which represents a potential threat to public health through the food chain, remains insufficiently investigated. Aim: The aim of this study was to characterize the AMR profiles of S. cohnii isolates from Chinese swine farms using integrated phenotypic and genotypic approaches. Methods: This study characterized 32 porcine-derived S. cohnii isolates from Jilin Province, China. Antimicrobial susceptibility testing against 20 antibiotics was performed using the agar dilution method, and the genomic basis of resistance was investigated through second-generation sequencing followed by comprehensive antibiotic resistance gene analysis. Results: Phenotypic testing revealed universal resistance (100%) to 11 antibiotics, including ampicillin and tetracycline. Genomic analysis identified 32 distinct resistance genes, with six of them (e.g., fusF, salE, vanTG) present in all isolates. Critically, multiple transferable multidrug-resistant genes were detected, including ermB (53.13%), ermC (18.75%), and cfrA (12.5%). Furthermore, silent glycopeptide resistance genes (e.g., vanTG, vanYB) were identified despite phenotypic susceptibility to vancomycin. Conclusion: This study establishes porcine S. cohnii as a significant reservoir of diverse and mobile resistance genes, including silent determinants with potential for horizontal transfer. These findings underscore the necessity of integrating genomic surveillance of commensal staphylococci in livestock within a One Health framework to mitigate the dissemination of resistance to human pathogens. Keywords: Staphylococcus cohnii, Antimicrobial resistance, Swine, Resistance genes, Second-generation sequencing analysis. IntroductionCoagulase-negative staphylococci (CoNS) represent a diverse group of Gram-positive bacteria characterized by their inability to produce coagulase (Becker et al., 2020; França et al., 2021). While generally regarded as less virulent than Staphylococcus aureus (S. aureus) and less frequently associated with severe human infections (Becker et al., 2020; Michels et al., 2021), their clinical significance in veterinary medicine continues to grow. CoNS are increasingly recognized as important pathogens in various livestock species, contributing to a range of infectious diseases with notable economic and welfare implications. In dairy cattle, CoNS are among the most frequently isolated bacteria associated with bovine mastitis, a condition that leads to substantial economic losses due to reduced milk yield and quality (Pyorala and Taponen, 2009). In swine, specific CoNS species, such as Staphylococcus hyicus and Staphylococcus sciuri, have been identified as causative agents of exudative epidermitis, a severe skin disease that particularly affects young piglets (Chen et al., 2007; Fišarová et al., 2019). CoNS have also been implicated in more invasive infections, such as osteomyelitis, in farm animals, underscoring their broader pathogenic potential beyond superficial and production-related diseases (Bonvegna et al., 2021). Their high prevalence and propensity to develop multidrug resistance make enhanced surveillance imperative. Staphylococcus cohnii, a member of the CoNS group, has been implicated in multiple human infections such as endocarditis, urinary tract infections, spontaneous bacterial peritonitis, and brain abscesses (Yamashita et al., 2005; Shahandeh et al., 2015; Motta et al., 2020; Lei et al., 2023). The clinical significance of this species is further heightened by the emergence of multidrug-resistant S. cohnii strains in agricultural environments, such as poultry and swine farms (Han et al., 2017; Bonvegna et al., 2021). Molecular studies provide a crucial link between these reservoirs and human health, having identified identical resistance genes, including mecA in both livestock-associated S. cohnii isolates and those from human clinical cases, suggesting potential transmission routes (Tulinski et al., 2012). This ecological connection is supported by findings of shared resistance determinants, such as novel fusidic acid resistance genes, unique SCCmec elements, and the multidrug resistance gene cfr, in S. cohnii from diverse sources (Zong and Lü, 2010; Chen et al., 2015; Gao et al., 2022). These findings collectively challenge the traditional view of S. cohnii as a benign commensal and underscore its potential role as a significant reservoir for resistance genes, with implications for public health through possible transmission via occupational exposure, the food chain, or environmental dissemination. This study was conducted in Jilin Province, a major swine-producing region in China. The province’s intensive farming practices and substantial swine population create an environment conducive to the development and spread of antimicrobial resistance (AMR). Investigating the resistance profiles of bacteria such as S. cohnii in such a representative area provides critical insights that are likely reflective of broader AMR trends within the Chinese livestock industry. This study aims to comprehensively characterize the AMR profiles of S. cohnii isolates from Chinese swine farms using integrated phenotypic and genotypic approaches. Our investigation provides current minimal inhibitory concentration (MIC) data, elucidates the genetic basis of AMR in this species, and establishes a foundation for future research on CoNS genomics and resistance mechanisms. Materials and MethodsBacterial strains and phylogenetic analysisA total of 32 strains of S. cohnii were isolated from 835 porcine nasal swab samples collected from large-scale intensive swine farms across multiple cities in Jilin Province, China. These farms, characterized by their intensive management practices and substantial herd sizes, represent the predominant swine production system in this major agricultural region. Sampling was conducted by trained veterinarians following standardized protocols under appropriate biosafety conditions. Nasal swabs were aseptically collected using sterile cotton swabs from pigs selected through a systematic sampling approach. Within pre-selected pens housing pigs with the target condition, sampling focused on grower-finisher pigs exhibiting mild respiratory symptoms (e.g., cough, nasal discharge, or increased respiratory rate). Any pig that had received antibiotic treatment within the 4 weeks prior to sampling was excluded to ensure that the observed resistance profiles were not directly confounded by recent therapy. The procedure involved gentle insertion of the swab into the anterior nares (approximately 2–3 cm in depth) with a brief, circular motion for sample collection, lasting less than 10 seconds per animal. Animals were minimally restrained by handlers to ensure safety and minimize stress. The collected swabs were immediately placed in Amies transport medium and maintained at 4°C during transport to the laboratory. Samples were inoculated onto Brain Heart Infusion (BHI) agar plates and incubated aerobically at 37°C using a Heratherm IGS60 incubator (Thermo Scientific). The incubation period of 12 hours was determined based on preliminary experiments and real-time monitoring of colonial growth to optimize the recovery of Staphylococcus species while minimizing overgrowth of other commensal flora. Subsequent isolation and purification procedures were performed for bacterial identification. Following primary isolation, all isolates were purified on BHI agar plates using the standard streaking technique to obtain single, well-isolated colonies. After incubation at 37°C for 12 hours, individual colonies exhibiting uniform morphology were selected and subcultured to ensure culture purity. Preliminary identification was performed through Gram staining, which confirmed that all isolates were Gram-positive staphylococci. The purified gram-positive staphylococcal isolates were then processed for subsequent molecular identification. Genomic DNA was extracted from pure bacterial cultures using the Bacterial Genomic DNA Rapid Extraction Kit (Aidlab Biotechnologies, China) following the manufacturer’s protocol. Strain identification was conducted through 16S rRNA gene sequencing. The quality of extracted DNA was evaluated by measuring concentration and purity (A260/A280 ratio) using a NanoDrop One spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and confirming integrity via 1% agarose gel electrophoresis. Amplification of the 16S rRNA gene was carried out using the universal primer pair 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) (Moreno et al., 2002). The thermal cycling protocol comprised an initial denaturation at 94°C for 5 minutes; 30 cycles of 95°C for 1 minute, 55°C for 1 minute, and 72°C for 1 minute; and a final extension at 72°C for 10 minutes. The resulting polymerase chain reaction amplicons were subsequently verified by 1% agarose gel electrophoresis to confirm the presence of a single band corresponding to the expected size (~1,500 bp) before proceeding to sequencing. Species identification was performed by 16S rRNA sequencing. DNA sequences were compared to genes in the National Center for Biotechnology Information (NCBI) GenBank using the Basic Local Alignment Search Tool (BLAST) algorithm. The phylogenetic tree was constructed based on 16S rRNA gene sequences, comprising 87 strains in total: 32 S. cohnii isolates obtained in this study and 55 reference strains retrieved from the NCBI database. The following criteria were used to select the reference strains: (1) source hosts (covering diverse origins); (2) geographic distribution (representing different regions worldwide); and (3) sequence quality (ensuring high-quality, near-full-length 16S rRNA sequences with minimum gaps). Phylogenetic reconstruction was performed using the Neighbor-Joining method implemented in Molecular Evolutionary Genetics Analysis version 11, with evolutionary distances computed using the Tamura-Nei model and bootstrap analysis based on 1,000 replicates. The resulting phylogenetic tree was visualized and annotated using tvBOT version 2.6.1 with the following configurations: circular phylogram layout, color-coded clades, and customized symbols highlighting strain origins. The use of 16S rRNA sequences alone has limited resolution for intraspecies differentiation, and future studies would benefit from incorporating whole-genome sequencing data for more precise phylogenetic analysis. Antimicrobial drug susceptibility testingPrior to antimicrobial susceptibility testing, all 32 S. cohnii isolates underwent verification of both purity and taxonomic identity. Fresh subcultures were prepared on BHI agar plates and incubated at 37°C for 12 hours. Colony morphology, size, and pigmentation were examined to assess culture purity. Following this, taxonomic assignment was further validated through 16S rRNA gene amplification and sequencing from single colonies, employing the primer sets and experimental procedures detailed in the section on strain identification. The agar dilution method was used to test the susceptibility of the isolated S. cohnii strains to the following 20 antibiotics: ampicillin (AMP), penicillin (PEN), cefoxitin (FOX), cefotaxime (CTX), meropenem (MEM), tetracycline (TET), rifampicin (RIF), sulfamethoxazole (SMZ), trimethoprim (TMP), fusidic acid (FUS), chloramphenicol (CHL), streptomycin (STR), linezolid (LZD), tigecycline (TGC), ciprofloxacin (CIP), mupirocin (MUP), gentamicin (GEN), vancomycin (VAN), erythromycin (ERY), and clindamycin (CLI). The abbreviations used for each antibiotic (indicated in parentheses) correspond to those employed in the subsequent data visualization figures. Bacteria were cultured in BHI broth for 6 hours, and the concentration of the bacterial suspension was adjusted to approximately 1 × 108 CFU/ml. Bacterial suspensions were inoculated equally spaced onto each plate using a multipoint inoculator, and each strain was tested in triplicate. All plates were incubated for 24 hours at 37°C. The MIC value recorded was the lowest antibiotic concentration that sufficiently inhibited visible bacterial growth. The clinical breakpoints for interpreting MIC values were based on the criteria for Staphylococcus spp. as defined by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) Breakpoint Tables (Version 14.0, 2024). Staphylococcus aureus ATCC 25923 was used as the quality control strain for susceptibility testing. The visualization and analysis of the MIC results were performed using Origin 2024 software. Predictive analysis of resistance genesPrior to genomic analysis, all 32 S. cohnii isolates underwent 16S rRNA sequencing to verify strain purity and identity, after which second-generation sequencing was performed. After cultivation in BHI broth at 37°C for 16 hours, bacterial cells were harvested by centrifugation. The resulting cell pellets were submitted to KAITAI-BIO (Hangzhou, China) for draft genome sequencing, which generated an average sequencing depth of 100× per genome. Libraries were constructed using the Enzymic Universal DNAseq Library Prep Kit (KAITAI-BIO, AT4107), and sequencing was carried out on the Illumina NovaSeq 6000 platform in PE150 mode. The raw reads underwent quality control filtering, and the resulting high-quality data were assembled de novo using Unicycler. The assembly quality was assessed based on the contig N50 values, and annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline. Antibiotic resistance genes were identified by analyzing the annotated genomes against the Comprehensive Antibiotic Resistance Database (CARD, version 4.0.1) using the Resistance Gene Identifier (RGI) tool (web portal version 6.0.5). The analysis followed a three-step workflow: open reading frames were initially predicted from genomic sequences using Prodigal; homology searches were subsequently performed with DIAMOND; and finally, stringent filtering was applied to retain only high-confidence hits meeting either “Perfect” (requiring 100% amino acid identity to CARD reference sequences) or “Strict” (with a bitscore ≥500) criteria for subsequent analysis. The resulting resistance gene profiles were visualized using OriginPro 2024. Ethical approvalThis study and its sampling protocol were reviewed and approved by the Committee of JiLin Agricultural Science and Technology College (Approval No. LLSC202301055). Nasal swab samples were collected from swine on farms with the verbal consent of the farm manager. The sampling procedure detailed in the Methods section was classified as a non-invasive, minimal-risk procedure by the ethics committee. This classification and subsequent approval were based on the following criteria: (a) the procedure involves only superficial mucosal contact without penetrating body cavities or causing tissue damage; (b) it is of very short duration; and (c) it is considered a routine animal husbandry practice analogous to standard health inspections, causing no harm or undue stress to the animals. All procedures were performed in accordance with relevant ethical standards and guidelines for the welfare of animals used in scientific research. Results and DiscussionBacterial strains and phylogenetic analysisThe taxonomic identity of all 32 strains was confirmed by 16S rRNA gene sequencing analysis using the NCBI BLAST, with all sequences exhibiting > 99.00% identity to reference sequences of S cohnii. The 16S rRNA gene sequences of all 32 S. cohnii isolates have been deposited in the NCBI database under the accession numbers listed in Table 1. Table 1. NCBI accession numbers for 16S rRNA sequences and second-generation sequencing data of S. cohnii isolates.
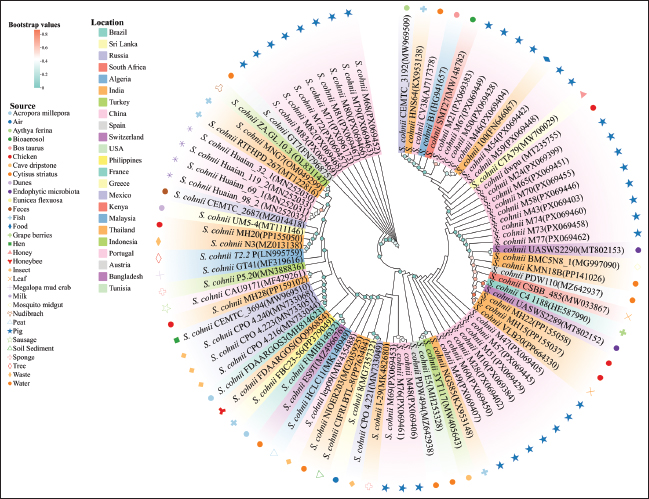
The phylogenetic analysis of 87 S. cohnii isolates—including 32 porcine-derived strains from Jilin Province, China, and 55 isolates from various host species and geographic regions—is presented in Figure 1. The results reveal considerable phylogenetic diversity among the 32 porcine strains, despite their common origin. Furthermore, these strains exhibit significant divergence from isolates originating in other regions.
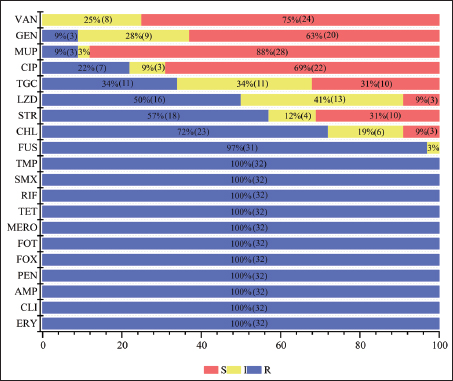
Fig. 1. Phylogenetic tree based on 16S rRNA sequences of the isolates and selected species. The phylogenetic tree comprised 87 32 of which were experimental strains from this study, isolated from samples obtained from Jilin Province, China. The remaining 55 strains were derived from 16S rRNA sequences of S. cohnii isolated from various species in disparate areas, sourced from the NCBI database. The base color of the leaf indicates the isolated species of the strains. The coloration of the circles on the branches shows the bootstrap values. The symbols displayed in the outermost circle indicate the region from which the strains were isolated. Multidrug resistance phenotypes of S. cohnii The AMR phenotypes of the 32 porcine-derived S. cohnii isolates are summarized in Figure 2. Based on the epidemiological cut-off values established by the EUCAST for S. aureus and CoNS, a non-wildtype phenotype was prevalent against multiple drug classes. High levels of resistance were observed to PENs, cephalosporins, carbapenems, TETs, and oxazolidinones. In contrast, susceptibility remained comparatively higher to aminoglycosides, glycopeptides, and macrolides.
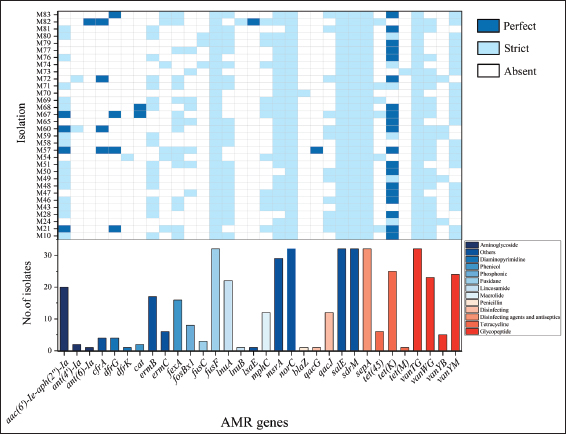
Fig. 2. AMR rates of S. cohnii to 20 antibiotics. This figure illustrates the drug resistance profile of 32 S. cohnii isolates originating from swine in Jilin Province, China. The blue section represents resistant isolates (R in the legend), yellow indicates intermediate (I in the legend), and red denotes susceptible (S in the legend). The abbreviations for antibiotics are as follows: AMP, PEN, FOX, CTX, MEM, TET, RIF, SMZ, TMP, FUS, CHL) STR, LZD, TGC, CIP, MUP, GEN, VAN, ERY, and CLI. The clinical breakpoints for interpreting MIC values were based on the criteria for Staphylococcus spp. as defined by the EUCAST Breakpoint Tables (Version 14.0, 2024). The percentage values are labeled with the corresponding number of isolates (n), with the value for the 3% bar (n=1) provided in the legend. All percentage values are rounded to the nearest whole number. All isolates (32/32, 100%) were resistant to 11 antibiotics, with the following MIC ranges for each: AMP (512 μg/ml), PEN (16–128 μg/ml), FOX (32–128 μg/ml), CTX (8–64 μg/ml), MERO (4–128 μg/ml), TET (16–128 μg/ml), RIF (1–128 μg/ml), SMX (512 μg/ml), TMP (512 μg/ml), ERY (64–512 μg/ml), and CLI (32–512 μg/ml). High resistance rates were observed against FUS (31/32, 97%) and CHL (23/32, 72%). In contrast, relatively high susceptibility was observed to GEN (20/32, 63%), CIP (22/32, 69%), VAN (24/32, 75%), and MUP (28/32, 88%). All interpretations were based on the breakpoints provided by the EUCAST Breakpoint Tables (Version 14.0, 2024). Prevalence of AMR genes in S. cohnii isolates Second-generation sequencing of 32 S. cohnii isolates led to the identification of 32 AMR genes (Fig. 3). Six of these genes—fusF, norC, salE, sdrM, sepA, and the vanT gene in the vanG cluster (vanTG)—were detected in all isolates (32/32, 100%). Although no novel resistance genes were identified, multiple genes mediating multidrug resistance were detected, including: cfrA, which confers resistance to phenicol, oxazolidinone, streptogramin, lincosamide, and pleuromutilin antibiotics (4/32, 12.50%); ermB (17/32, 53.13%) and ermC (6/32, 18.75%), both mediating resistance to streptogramin, lincosamide, and macrolide antibiotics; lsaE (1/32, 3.13%) and salE (32/32, 100%), each conferring resistance to pleuromutilin, streptogramin, and lincosamide antibiotics; and msrA (29/32, 90.63%), which mediates resistance to streptogramin, lincosamide, and macrolide antibiotics.
Fig. 3. The AMR gene carriage profiles of 32 S. cohnii isolates. The upper heatmap section displays the resistance genes carried by each isolate, with the vertical axis indicating the names of individual strains. Dark blue represents “Perfect” hits indicating exact sequence matches to a resistance gene, light blue denotes “Strict” hits reflecting stringent but not exact matches, and white indicates “Absent” for genes not detected in a particular strain. The lower bar chart section annotates the types of antibiotics to which each gene confers resistance. The corresponding resistance phenotypes are annotations based on the CARD database. The horizontal axis lists the 32 identified resistance genes, while the vertical axis shows the number of isolates in which each gene was detected. Note: “vanTG” refers to the vanT gene in the vanG cluster, “vanWG” to the vanW gene in the vanG cluster, “vanYB” to the vanY gene in the vanB cluster, and “vanYM” to the vanY gene in the vanM cluster. The “Others” category in the legend represents genes that confer resistance to two or more classes of antibiotics. All resistance genes listed confer resistance to antimicrobial antibiotics. Disinfectants and antiseptics were not experimentally evaluated in this study due to the current lack of standardized susceptibility testing methods and breakpoints for S. cohnii. RationaleIn this study, the analysis of AMR in porcine-derived S. cohnii revealed a substantial reservoir of genes capable of mediating MDR. The selection of porcine-derived S. cohnii for this investigation was based on several critical considerations: (1) swine represent a major reservoir of AMR bacteria due to extensive antibiotic use in intensive farming; (2) S. cohnii, though often overlooked as a commensal organism in animal hosts, may serve as a potential contributor to gene exchange in microbial communities; and (3) the potential transmission of resistant strains through the food chain poses significant public health concerns. As a coagulase-negative Staphylococcus, S. cohnii is often overlooked in animal hosts. Previous research has demonstrated that S. cohnii can undergo adaptive evolution through genetic exchange with other bacterial species under antibiotic-selective pressure (Szewczyk et al., 2003). The following sections will analyze the characteristics of AMR in S. cohnii isolates from swine in Jilin Province, China, from several key perspectives. Phenotypic resistance profiles and implications for swine healthThe high prevalence of multidrug-resistant S. cohnii isolates observed in this study reveals critical insights into AMR dynamics within swine production systems. The extensive resistance to PENs, cephalosporins, and TETs—antibiotics commonly employed in swine production—suggests potential limitations in therapeutic efficacy for staphylococcal infections. This is particularly concerning given that all isolates exhibited resistance to 11 different antimicrobial agents, indicating substantial selective pressure from antibiotic use in the regional swine industry. The persistence of such resistant strains may lead to treatment failures, increased mortality rates, and economic losses due to reduced productivity and higher veterinary costs. Genotypic characterization of resistance determinantsOur findings identified a broad spectrum of AMR genes conferring resistance to 11 classes of antibiotics, with a particularly high prevalence (20/32 isolates) of the bifunctional aminoglycoside resistance gene aac(6’)-Ie-aph(2’’)-Ia. This gene encodes an enzyme capable of inactivating virtually all clinically relevant aminoglycosides, including GEN, through dual acetylation and phosphorylation mechanisms, thereby posing a significant challenge to therapeutic efficacy and future drug development (Daigle et al., 1999). Moreover, we observed a complex genetic framework underlying resistance to macrolides, lincosamides, and streptogramin B (MLSB). Specifically, the ermB and ermC genes, which confer cross-resistance by methylating the 23S rRNA target site, were frequently detected. This primary mechanism was further supported by the concomitant presence of the macrolide-efflux gene msrA and the lincosamide-inactivating genes lnuA and lnuB. The co-occurrence of these genes indicates a synergistic interaction, collectively reinforcing a robust and multifaceted MLSB resistance phenotype in the studied S. cohnii population. These findings not only validate the role of these genes in observed resistance but also highlight the potential for co-selection and persistence of MLSB resistance in swine-associated staphylococci. Potential for genetic exchange and resistance disseminationFurthermore, our findings demonstrate that S. cohnii carries a diverse array of AMR genes within the swine microbiome. The detection of multiple resistance determinants, including cfrA, ermB, ermC, and msrA, suggests the theoretical potential for genetic exchange within the swine production environment, although our study did not determine the specific genetic contexts of these elements. It should be noted that genes such as blaZ, ermC, and tet(K) are frequently reported to be associated with plasmids or other mobile genetic elements (MGEs) in staphylococc, though our methodology cannot confirm their genetic location in these isolates. The universal presence of fusF, norC, and salE among all isolates suggests these genes may represent core resistance elements in porcine-derived S. cohnii populations. The coexistence of multiple resistance mechanisms creates a genetic environment that could maintain and potentially disseminate resistance traits under continued antimicrobial selection pressure. Silent resistance genes and public health implicationsOf particular concern was the detection of genes from various van clusters, such as vanTG, vanWG, vanYB, and vanYM, which are known to mediate resistance to glycopeptide antibiotics. Interestingly, the MIC results indicated that all isolates remained phenotypically susceptible to VAN. This discrepancy suggests that these resistance genes may be silent or not fully expressed in the current genetic background of our S. cohnii isolates. However, their presence constitutes a significant latent risk. Although the short-read sequencing data used in this study preclude the precise determination of the genetic context of these van genes, their reported linkage to MGEs in other staphylococcal species suggests a potential for horizontal gene transfer (HGT). The vanG operon, for instance, has been reported to be located on transferable genetic elements that can also carry the ermB gene—a gene concurrently detected in our study. This genetic linkage, if present, raises the possibility of co-selection under antimicrobial pressure, wherein the use of macrolides (selecting for ermB) could inadvertently promote the spread of the linked VAN resistance genes. Future investigations employing long-read sequencing technologies are warranted to conclusively determine whether these silent van genes are located on plasmids or other MGEs in our isolates. Mechanistically, these genes confer resistance through sophisticated modifications of the peptidoglycan precursor termini. VanT, a membrane-bound serine racemase, produces D-serine, which is incorporated into a D-Ala-D-Ser terminus by VanC, reducing VAN binding affinity. Alternatively, the D,D-carboxypeptidase activity of VanY removes the terminal D-Ala, facilitating the formation of a D-Ala-D-Lac terminus with even lower affinity for VAN. Although the function of the accessory protein VanW remains unclear, its presence is characteristic of certain resistance operons. Therefore, the silent van genes identified in this study represent a potential public health concern. Their potential mobilization from S. cohnii into a more virulent background, such as MRSA, could theoretically activate their expression, underlining the need for surveillance (Depardieu et al., 2003). The silent resistance reservoir in animal-associated bacteria underscores the need for ongoing surveillance and further research into the mobility of glycopeptide resistance genes to preempt future clinical crises. In addition, we identified other clinically crucial determinants: the PhLOPSA-conferring cfrA (12.5%) threatening last-resort LZD, and lsaE contributing to universal CLI resistance. The coexistence of multiple resistance mechanisms, including ribosomal protection and enzymatic inactivation, creates compounded resistance effects that complicate treatment options (Li et al., 2013; Candela et al., 2017). Contribution to AMR surveillanceThese findings provide critical baseline data for China’s national AMR surveillance program, particularly within the swine production sector. The comprehensive resistance profiles and genetic characterization of S. cohnii from Jilin Province—a major swine-producing region—offer valuable insights into the distribution of resistance mechanisms in livestock-associated staphylococci. The cfrA gene, which confers cross-resistance to five antibiotic classes, including the critically important LZD, represents an early warning for public health authorities. Our study underscores the necessity of integrating commensal bacteria such as S. cohnii into ongoing AMR monitoring programs, as they may serve as indicators of emerging resistance trends. The phylogenetic evidence of regional divergence further emphasizes the need for location-specific surveillance strategies that account for local antimicrobial usage patterns and farming practices. Regional and national epidemiological contextThe high prevalence of multidrug-resistant S. cohnii uncovered in this study from Jilin Province’s swine farms underscores a significant local epidemiological concern. The intensive animal husbandry practices in this major agricultural region undoubtedly exert a strong selective pressure, fostering the persistence and dissemination of resistant strains. While our findings highlight a serious issue within Jilin, it is crucial to consider whether this is part of a broader national pattern. Although direct, large-scale surveillance data on S. cohnii from other Chinese regions are still limited, emerging evidence suggests that this pathogen is indeed a concern beyond our study area. Notably, a recent report by Dong et al. (2024)documented the first isolation of a highly pathogenic and multidrug-resistant S. cohnii strain from a wildlife species (Myocastor coypus) in Hunan Province, Southern China. Although direct evidence of S. cohnii transmission across China remains limited, the identification of a pathogenic and multidrug-resistant strain in Hunan Province, a region geographically and ecologically distinct from our study area in Jilin, provides a crucial indication that the threat of this bacterium may extend beyond a single locality. The presence of strains in both northern and southern China, albeit in different host species, hints at a potentially wider, yet uncharacterized, circulation of S. cohnii. Given the interconnectedness of modern livestock and trade networks, the dissemination of these resistant strains or their genetic determinants is a plausible risk. Therefore, our findings in Jilin, viewed alongside the report from Hunan, highlight the need for heightened awareness and suggest that expanded surveillance is warranted to assess the true national scope of S. cohnii as a public health concern. Study limitationsThis study has several limitations that should be considered when interpreting the results. First, the analysis of 32 S. cohnii isolates, while providing valuable insights, represents a limited sample size that may affect the generalizability of our findings to broader geographic regions or different farming systems. Second, we were unable to obtain detailed farm-level data on operation size, management practices, and antimicrobial usage due to inconsistencies in record-keeping across multiple production sites and cycles. The inclusion of such epidemiological data would have strengthened the contextual interpretation of the observed resistance patterns. Third, the use of second-generation sequencing technology, while effective for identifying the presence of resistance genes, precludes the precise determination of their genetic context—specifically, whether these genes are located on chromosomes, plasmids, or other MGEs. This limitation restricts our ability to definitively assess the horizontal transfer potential of the identified resistance determinants. Future studies incorporating long-read sequencing technologies and systematic collection of farm-level metadata would provide greater resolution of resistance gene mobility and enhance our understanding of the drivers of AMR dissemination in swine production systems. Fourth, this study did not evaluate resistance to disinfectants or antiseptics, as standardized methods for S. cohnii are lacking and interpretive criteria. Future studies should incorporate such testing when validated guidelines become available to provide a more comprehensive understanding of S. cohnii resistance in farm environments. ConclusionThis study characterized 32 S. cohnii isolates from swine in Jilin Province, China. Antimicrobial susceptibility testing revealed high resistance rates to multiple drug classes, including complete resistance (100%) to 11 antibiotics. Genomic analysis identified 32 resistance genes, including cfrA, ermB/C, and silent van clusters. The presence of these genes, some of which are frequently reported on MGEs in other staphylococci, highlights that porcine S. cohnii may represent a potential reservoir for multidrug resistance determinants. These findings establish porcine S. cohnii as a significant reservoir of transmissible multidrug resistance. From a One Health perspective, proactive surveillance of commensal staphylococci in livestock is essential to mitigate the potential dissemination of these resistance determinants to human pathogens. To manage this risk, implementing enhanced biosecurity protocols on farms to reduce bacterial load, alongside routine monitoring of resistance gene prevalence in animal and environmental samples, is strongly recommended. AcknowledgmentsWe extend our appreciation to the Jilin Province Science and Technology Innovation Platform Subsidy Project and the staff of the Jilin Agricultural Science and Technology College. Conflict of interestThe authors declare that they have no conflicts of interest to the work reported in this paper. FundingThe funding for this research was provided by Jilin Province Science and Technology Innovation Platform Subsidy Project (Grant number YDZJ202302CXJD038). Authors’ contributionsMethodology, H. N. Li; Software, M. R. Ma; Validation, N. Xu and M. R. Ma; Investigation, Z. L. Yang and S. J. Song; Resources, Z. L. Yang; Visualization, B. L. Sui; Writing – Original Draft Preparation, N. Xu and M. R. Ma; Writing – Review & Editing, W. L. Sha, B. S. Yin and W. L. Dong; Supervision, W. L. Sha, B. S. Yin and W. L. Dong; Funding Acquisition, W. L. Dong. Data availabilityThe 16S rRNA gene sequences and Second-generation sequencing data of all 32 S. cohnii isolates have been deposited in the NCBI database. The corresponding accession numbers are provided in Table 1 of this manuscript. Relevant data supporting the findings are available from the corresponding author upon reasonable request. ReferencesBecker, K., Both, A., Weißelberg, S., Heilmann, C. and Rohde, H. 2020. Emergence of coagulase-negative staphylococci. Expert. Rev. Anti. Infect. Ther. 18(4), 349–366. Bonvegna, M., Grego, E., Sona, B., Stella, M.C., Nebbia, P., Mannelli, A. and Tomassone, L. 2021. Occurrence of Methicillin-Resistant Coagulase-Negative Staphylococci (MRCoNS) and Methicillin-Resistant Staphylococcus aureus (MRSA) from pigs and farm environment in Northwestern Italy. Antibiotics (Basel) 10(6), 676. Candela, T., Marvaud, J.C., Nguyen, T.K. and Lambert, T. 2017. A cfr-like gene cfr(C) conferring linezolid resistance is common in Clostridium difficile. Int. J. Antimicrob. Agents. 50(3), 496–500. Chen, H.J., Hung, W.C., Lin, Y.T., Tsai, J.C., Chiu, H.C., Hsueh, P.R. and Teng, L.J. 2015. A novel fusidic acid resistance determinant, fusF, in Staphylococcus cohnii. J. Antimicrob. Chemother. 70(2), 416–419. Chen, S., Wang, Y., Chen, F., Yang, H., Gan, M. and Zheng, S.J. 2007. A highly pathogenic strain of Staphylococcus sciuri caused fatal exudative epidermitis in piglets. PLos One 2(1), 147. Daigle, D.M., Hughes, D.W. and Wright, G.D. 1999. Prodigious substrate specificity of AAC(6’)-APH(2’), an aminoglycoside antibiotic resistance determinant in enterococci and staphylococci. Chem. Biol. 6(2), 99–110. Depardieu, F., Bonora, M.G., Reynolds, P.E. and Courvalin, P. 2003. The vanG glycopeptide resistance operon from Enterococcus faecalis revisited. Mol. Microbiol. 50(3), 931–948. Dong, W., Peng, N., Yang, L., Ning, H., Fan, J., Li, X., Chen, Y., Han, X. and Ge, M. 2024. First report of Myocastor coypus Infected with Staphylococcus cohnii. Transbound. Emerg. Dis. 1(1), 3710299. Fišarová, L., Pantůček, R., Botka, T. and Doškař, J. 2019. Variability of resistance plasmids in coagulase-negative staphylococci and their importance as a reservoir of antimicrobial resistance. Res. Microbiol. 170(2), 105–111. França, A., Gaio, V., Lopes, N. and Melo, L.D.R. 2021. Virulence factors in coagulase-negative Staphylococci. Pathogens 10(2), 170. Gao, Y., Wang, Z., Fu, J., Cai, J., Ma, T., Xie, N., Fan, R., Zhai, W., Feßler, A.T., Sun, C., Wu, C., Schwarz, S., Zhang, R. and Wang, Y. 2022. Spreading of cfr-carrying plasmids among staphylococci from humans and animals. Microbiol. Spectr. 10(6), 246122; e0246122 Han, J.E., Lee, S., Jeong, D.G., Yoon, S.W., Kim, D.J., Lee, M.S., Kim, H.K., Park, S.K., Kim, J.H. and Park, S.C. 2017. Complete genome sequence of multidrug-resistant Staphylococcus cohnii ssp. urealyticus strain SNUDS-2 isolated from farmed duck, Republic of Korea. J. Glob. Antimicrob. Resist. 10, 37–39. Lei, Y., Guo, Q., Liu, J., Huang, H. and Han, P. 2023. Staphylococcus cohnii infection diagnosed by metagenomic next generation sequencing in a patient on hemodialysis with cirrhotic ascites: a case report. Front. Cell. Infect. Microbiol. 13, 1240283. Li, B., Wendlandt, S., Yao, J., Liu, Y., Zhang, Q., Shi, Z., Wei, J., Shao, D., Schwarz, S., Wang, S. and Ma, Z. 2013. Detection and new genetic environment of the pleuromutilin-lincosamide-streptogramin A resistance gene lsa(E) in methicillin-resistant Staphylococcus aureus of swine origin. J. Antimicrob. Chemother. 68(6), 1251–1255. Michels, R., Last, K., Becker, S.L. and Papan, C. 2021. Update on coagulase-negative staphylococci-what the clinician should know. Microorganisms 9(4), 830. Moreno, C., Romero, J. and Espejo, R.T. 2002. Polymorphism in repeated 16S rRNA genes is a common property of type strains and environmental isolates of the genus Vibrio. Microbiol (Reading) 148(Pt 4), 1233–1239. Motta, J.C., Forero-Carreño, C., Arango, A. and Sánchez, M. 2020. Staphylococcus cohnii endocarditis in native valve. New. Microbes. New. Infect. 38, 100825. Pyorala, S. and Taponen, S. 2009. Coagulase-negative staphylococci-emerging mastitis pathogens. Vet. Microbiol. 134(1-2), 3–8. Shahandeh, Z., Shafi, H. and Sadighian, F. 2015. Association of Staphylococcus cohnii subspecies urealyticum infection with recurrence of renal staghorn stone. Caspian J. Intern. Med. 6(1), 40–42. Szewczyk, E.M., Nowak, T., Cieślikowski, T. and Różalska, M. 2003. Potential role of Staphylococcus cohnii in a hospital environment. Microb. Ecol. 15(1), 51–56. Tulinski, P., Fluit, A.C., Wagenaar, J.A., Mevius, D., Van De Vijver, L. and Duim, B. 2012. Methicillin-resistant coagulase-negative staphylococci on pig farms as a reservoir of heterogeneous Staphylococcal cassette chromosome Mec elements. Appl. Environ. Microbiol. 78(2), 299–304. Yamashita, S., Yonemura, K., Sugimoto, R., Tokunaga, M. and Uchino, M. 2005. Staphylococcus cohnii as a cause of multiple brain abscesses in Weber-Christian disease. J. Neurol. Sci. 238(1-2), 97–100. Zong, Z. and Lü, X. 2010. Characterization of a new SCCmec element in Staphylococcus cohnii. PLos One 5(11), e14016. | ||
| How to Cite this Article |
| Pubmed Style Xu N, Ma M, Li H, Yang Z, Sui B, Song S, Sha W, Yin B, Dong W. Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, China. Open Vet. J.. 2026; 16(1): 711-720. doi:10.5455/OVJ.2026.v16.i1.66 Web Style Xu N, Ma M, Li H, Yang Z, Sui B, Song S, Sha W, Yin B, Dong W. Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, China. https://www.openveterinaryjournal.com/?mno=289279 [Access: June 22, 2026]. doi:10.5455/OVJ.2026.v16.i1.66 AMA (American Medical Association) Style Xu N, Ma M, Li H, Yang Z, Sui B, Song S, Sha W, Yin B, Dong W. Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, China. Open Vet. J.. 2026; 16(1): 711-720. doi:10.5455/OVJ.2026.v16.i1.66 Vancouver/ICMJE Style Xu N, Ma M, Li H, Yang Z, Sui B, Song S, Sha W, Yin B, Dong W. Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, China. Open Vet. J.. (2026), [cited June 22, 2026]; 16(1): 711-720. doi:10.5455/OVJ.2026.v16.i1.66 Harvard Style Xu, N., Ma, . M., Li, . H., Yang, . Z., Sui, . B., Song, . S., Sha, . W., Yin, . B. & Dong, . W. (2026) Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, China. Open Vet. J., 16 (1), 711-720. doi:10.5455/OVJ.2026.v16.i1.66 Turabian Style Xu, Nuo, Mingrui Ma, Hannuo Li, Zhuolei Yang, Binglun Sui, Shaojie Song, Wanli Sha, Baishuang Yin, and Wenlong Dong. 2026. Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, China. Open Veterinary Journal, 16 (1), 711-720. doi:10.5455/OVJ.2026.v16.i1.66 Chicago Style Xu, Nuo, Mingrui Ma, Hannuo Li, Zhuolei Yang, Binglun Sui, Shaojie Song, Wanli Sha, Baishuang Yin, and Wenlong Dong. "Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, China." Open Veterinary Journal 16 (2026), 711-720. doi:10.5455/OVJ.2026.v16.i1.66 MLA (The Modern Language Association) Style Xu, Nuo, Mingrui Ma, Hannuo Li, Zhuolei Yang, Binglun Sui, Shaojie Song, Wanli Sha, Baishuang Yin, and Wenlong Dong. "Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, China." Open Veterinary Journal 16.1 (2026), 711-720. Print. doi:10.5455/OVJ.2026.v16.i1.66 APA (American Psychological Association) Style Xu, N., Ma, . M., Li, . H., Yang, . Z., Sui, . B., Song, . S., Sha, . W., Yin, . B. & Dong, . W. (2026) Antimicrobial resistance and genomic analysis of Staphylococcus cohnii isolates from swine in Jilin, China. Open Veterinary Journal, 16 (1), 711-720. doi:10.5455/OVJ.2026.v16.i1.66 |

