




| Research Article | ||
Open Vet. J.. 2026; 16(2): 1213-1221 Open Veterinary Journal, (2026), Vol. 16(2): 1213-1221 Research Article Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, ChinaHannuo Li1#, Xiaorui Pan1#, Nuo Xu1, Guangyan Zhang1, Wanli Sha1,2,3, Baishuang Yin1,2,4, Wenlong Dong1,2,3,*1College of Animal Science and Technology, JiLin Agricultural Science and Technology College, Jilin, China 2JiLin Provincial Key Laboratory of Preventive Veterinary Medicine, Jilin, China 3JiLin Province Technology Innovation Center of Pig Ecological Breeding and Disease Prevention and Control, Jilin, China 4JiLin Province Cross Regional Cooperation Technology Innovation Center of Porcine Main Disease Prevention and Control, Jilin, China #Both authors have made the same contribution to this paper; they are cofirst authors. *Corresponding Author: Wenlong Dong. Animal Science and Technology College, JiLin Agricultural Science and Technology College Jilin, China. Email: dongwenlong888 [at] jlnku.edu.cn Submitted: 17/09/2025 Revised: 06/01/2026 Accepted: 20/01/2026 Published: 28/02/2026 © 2026 Open Veterinary Journal
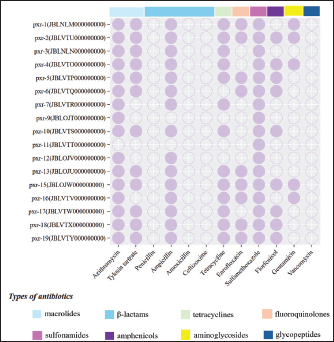
ABSTRACTBackground: Streptococcus pluranimalium is an emerging zoonotic pathogen. Its overall low isolation rate has contributed to the paucity of available data. However, data on its antimicrobial resistance (AMR) profile, particularly from swine in China, remain scarce. The escalating global challenge of AMR underscores the urgent need to elucidate resistance mechanisms in zoonotic veterinary pathogens. Aim: To the best of our knowledge, this is the first comprehensive characterization of AMR phenotypes and genotypes in S. pluranimalium isolates from diseased pigs in China, with an emphasis on identifying novel resistance gene combinations and evaluating their potential for cross-species dissemination. Methods: The AMR profile of 17 S. pluranimalium isolates was determined using minimum inhibitory concentration testing against a panel of antimicrobials. Next-generation sequencing was used to comprehensively investigate the genetic determinants of resistance, and a comparative analysis was conducted incorporating 5 publicly available genomes. Results: All 17 isolates were multidrug-resistant. They exhibited high resistance rates to sulfamethoxazole (100%), azithromycin (94.12%), tylosin tartrate (70.59%), and tetracycline (76.47%) but remained susceptible to penicillin, amoxicillin, ceftizoxime, and vancomycin. Genomic analysis identified 20 antimicrobial resistance genes. Notably, we report for the first time the co-occurrence of five aminoglycoside resistance genes (aad(6), aph(3')-IIIa, sat-4, aph(2'')-Ia, and patB) in this species, along with the simultaneous detection of lnuB and lnuC. Conclusion: Our next-generation sequencing analysis identified diverse and novel combinations of antibiotic resistance genes in porcine-derived S. pluranimalium, positioning it as a potential reservoir for AMR dissemination. These findings offer critical molecular insights into the AMR landscape in Chinese swine, emphasizing the risk of cross-species transmission and the need for integrated surveillance of this pathogen. Keywords: Antimicrobial resistance, Streptococcus pluranimalium, Pig. IntroductionThe Streptococcus genus consists of Gram-positive, catalase-negative cocci that are widespread and can cause infections in both humans and animals. These bacteria primarily colonize the respiratory, gastrointestinal, and urinary tracts. More than 70 species have been identified within the genus (Malke et al., 2018), many of which are zoonotic pathogens associated with severe diseases and high mortality rates. The escalating antimicrobial resistance (AMR) crisis poses a formidable challenge to global public health and sustainable livestock production. The overuse and misuse of antibiotics have exerted immense selective pressure, accelerating the evolution of resistant bacteria. This is particularly concerning for streptococci, which exhibit a pronounced capacity to acquire and horizontally transfer resistance genes through mobile genetic elements, leading to MDR that renders standard therapies ineffective. Genomic studies have been pivotal in uncovering the reservoirs and dissemination mechanisms of AMR genes in these pathogens (Wang et al., 2023). Within this context, characterizing the AMR profiles of emerging zoonotic agents, such as Streptococcus pluranimalium, is not merely an academic exercise but a critical necessity for developing effective surveillance and control strategies under the One Health framework. Streptococcus pluranimalium, a long-chain coccoid bacterium, was first described. It has been implicated in bovine mastitis, avian respiratory infections (Devriese et al., 1999), meningitis in calves, valvular endocarditis and septicemia in chickens, respiratory disease in dogs, and septicemia in fish (Kalhoro et al., 2015; Osman et al., 2013). Although initially considered an animal pathogen, S. pluranimalium was first isolated from humans in 2014 and has since been associated with severe conditions, including brain abscesses, endocarditis, and respiratory infections (Ahmed et al., 2018). These findings highlight the zoonotic potential of the species and its emerging threat to public health. Despite its broad host range, most studies have focused on bovine isolates, with limited attention given to swine-derived S. pluranimalium. To date, no studies have reported the isolation or AMR characteristics of S. pluranimalium from pigs in Jilin Province, China, highlighting a critical geographical and host-specific research gap. This study aimed to isolate and identify bacterial pathogens from diseased pigs from 11 pig farms in Jilin, China. To the best of our knowledge, this is the first study to isolate and characterize S. pluranimalium from pigs in this region. The recovered S. pluranimalium isolates were characterized through a combination of phenotypic and genotypic approaches. Phenotypically, antimicrobial susceptibility testing was performed via the agar dilution method to determine the minimum inhibitory concentration (MIC) against a panel of 12 clinically relevant antimicrobials, encompassing multiple classes (e.g., β-lactams, macrolides, tetracyclines, and sulfonamides). Genotypically, we performed genome sequencing based on next-generation sequencing technology for all isolates. The generated high-quality genomic data were used to comprehensively screen for known antimicrobial resistance genes (ARGs) and explore the genetic context of identified determinants. Furthermore, a comparative genomic analysis was performed by integrating additional publicly available S. pluranimalium genomes to contextualize the genetic diversity, ARG profiles, and potential for horizontal gene transfer of our isolates within a broader geographical and host-associated framework. The findings of this study aim to support the rational use of antibiotics in treating S. pluranimalium infections, thereby reducing economic impacts on swine production, and provide a foundation for understanding the molecular mechanisms and dissemination potential of antimicrobial resistance in this emerging pathogen. Materials and MethodsIsolation and identification of bacteriaThis study involved the aseptic collection of 1,224 fresh nasal samples from pigs across 11 distinct farms in Jilin Province, China, targeting those with mild respiratory disease identified by signs such as nasal discharge, coughing, or tachypnea. Following collection, nasopharyngeal swabs were placed in sterile tubes, stored at 4°C, and processed within 12 hours; they were then cultured on brain heart infusion (BHI) agar (Haibo Biotechnology Co., Qingdao, China) and incubated at 37°C for 12 hours to maximize pathogen recovery, with this temperature selected to match the physiological conditions of the porcine host. Gram staining of these isolates revealed Gram-positive cocci in chains. A total of 65 presumptive streptococcal isolates were obtained for molecular identification. The isolation of genomic DNA was performed on pure bacterial cultures using the Bacterial Genomic DNA Rapid Extraction Kit (Aidlab Biotechnologies, China), following the protocol specified for Gram-positive bacteria. The initial step consisted of lysing the bacterial cells with a proprietary buffer (Solution GY) along with Proteinase K at 70°C. Subsequently, the liberated DNA was bound to a silica membrane column by introducing a binding buffer (Solution CB) with isopropanol. Following the binding step, the DNA-membrane complex was washed twice to remove contaminants. Then, pure genomic DNA was recovered by elution into a preheated elution buffer. The concentration and purity (determined by the A260/A280 ratio) of the eluted DNA were quantified using a NanoDrop One spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Furthermore, the integrity of the extracted DNA was assessed using 1% agarose gel electrophoresis. Strain identification was conducted by sequencing the 16S rRNA gene using the primer pair 27F (5′-AGAGTTTGATCCTGGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) for upstream and downstream amplification, respectively (Moreno et al., 2002). PCR amplification was performed using a Biosafer-9702 Gradient PCR Thermal Cycler (BioSafer, China). The initial denaturation step was performed at 94°C for 5 minutes, followed by 30 cycles of denaturation at 95°C for 1 minute, annealing at 55°C for 1 minute, and extension at 72°C for 1 minute, with a final extension phase at 72°C for 10 minutes. Subsequent BLAST analysis against the National Center for Biotechnology Information database confirmed 17 of the 65 isolates as S. pluranimalium (with sequence identity ≥98.87%), which constituted the final collection for this study. In accordance with modern bacterial taxonomy, which prioritizes genomic data for accurate species assignment, no additional biochemical profiling was performed beyond Gram staining. Antimicrobial susceptibility testingThe MICs of the 17 S. pluranimalium isolates against 12 antimicrobial agents were determined by the agar dilution method, following the Clinical and Laboratory Standards Institute guidelines (M100-Ed33). The agents tested were azithromycin, tylosin tartrate, penicillin, ampicillin, amoxicillin, cefuroxime, tetracycline, enrofloxacin, sulfamethoxazole, florfenicol, gentamicin, and vancomycin. Bacterial suspensions were prepared by inoculating isolates into BHI broth and incubating them at 37°C for 8–12 hours in an air bath constant temperature shaker (THZ-98C). The main steps of agar dilution for antimicrobial susceptibility testing consist of adjusting the drug concentration to 0.03 μg/ml- 512 μg/ml (twofold dilution method: 0.03 μg/ml,0.06 μg/ml,0.125 μg/ml,0.25 μg/ml, and so on, up to 512 μg/ml). Staphylococcus aureus ATCC 25923 and Streptococcus pneumoniae ATCC 49619 were used as the quality control strains. The MIC results were interpreted as Susceptible (S), Intermediate (I), or Resistant (R) based on the CLSI breakpoints. For antimicrobials lacking specific breakpoints for S. pluranimalium, breakpoints for phylogenetically related species (e.g., S. suis for beta-lactams, viridans group streptococci for others) were applied as recommended by CLSI guidelines (M100-Ed33). Second-generation sequencing analysis and resistance gene predictionFor genomic DNA extraction, bacterial cultures were grown in BHI broth at 37°C with shaking for 8–12 hours at 200 rpm. Bacterial cultures were grown under controlled incubation conditions and harvested during the mid-logarithmic growth phase, typically after 12–14 hours of incubation, corresponding to an OD600 of approximately 0.5. Cells were pelleted by centrifugation at 10,000 × g for 5 minutes. Subsequent second-generation sequencing was performed by Bemac Biotechnology Co. (Qingdao, China). High-quality reads were filtered and then assembled de novo using SPAdes v3.6.2 to reconstruct the genomic sequences. Based on the Comprehensive Antibiotic Resistance Database (CARD v3.1.0, https://card.mcmaster.ca) and the Resistance Gene Identifier (RGI v5.1.1), the ARGs (azithromycin, tylosin tartrate, penicillin, ampicillin, amoxicillin, cefuroxime, tetracycline, enrofloxacin, sulfamethoxazole, florfenicol, gentamicin, and vancomycin) carried by 22 S. pluranimalium isolates were predicted using a stringent screening strategy that included both perfect/strict hits and loose hits (with ≥95% identity) nudged to strict. A total of 20 distinct antimicrobial resistance genes were identified and analyzed in this study (Table 1). Heatmaps were generated using TBtools (Toolbox for Biologists) software, version 2.337 (Chen et al., 2023). Comparative genomic analysis and public genome analysisA comparative genomic analysis was conducted to contextualize the genetic diversity and resistance gene profiles of our 17 porcine isolates within a broader framework. This analysis incorporated 5 publicly available S. pluranimalium genomes retrieved from the National Center for Biotechnology Information database. These strains were selected to provide geographical and source diversity and because preliminary screening indicated that they shared key AMR genes of interest with our isolates, allowing for a more robust analysis of AMR gene dissemination. The details of these public strains are as follows: SP28 (Source: Swine, Shandong, China), SP21-2 (Source: Environment/Food, Sichuan, China), Colony612 (Source: Environment/Food, Thailand), 14A0014 (Source: Clinical/Host-associated, Switzerland), and TH11417 (Source: Clinical/Host-associated, Henan, China). ResultsIsolation and genomic characterization of S. pluranimalium isolatesA total of 17 S. pluranimalium isolates were successfully obtained from 1,224 clinical swab samples collected from diseased pigs across 11 farms in Jilin Province, China, between May and October 2024, yielding an isolation rate of 1.39%. The taxonomic identity of all 17 isolates was confirmed through 16S rRNA gene sequencing, with all sequences exhibiting ≥ 98.87% identity to the reference sequence of S. pluranimalium upon analysis using the NCBI BLAST tool. The complete genome sequences of these isolates have been deposited in GenBank under the accession numbers listed in Figure 1. These 17 isolates formed the basis of all subsequent analyses. Multidrug resistance phenotypes of Streptococcus pluranimaliumThe phenotypic antibiotic resistance profiles of 17 S. pluranimalium isolates obtained from porcine hosts, along with 5 additional S. pluranimalium strains, are summarized in Figure 1. All isolates exhibited complete resistance (100.00%) to the sulfonamide antibiotic sulfamethoxazole. High resistance rates were observed against macrolides: azithromycin (94.12%) and tylosin tartrate (70.59%), as well as against the tetracycline antibiotic tetracycline (76.47%). Moderate resistance was detected against enrofloxacin (a fluoroquinolone) and florfenicol (an amphenicol), both with a resistance rate of 52.94%. Notably, while the isolates showed (100%) susceptibility to three β-lactam antibiotics—penicillin, amoxicillin, and ceftizoxime—a high resistance rate (88.24%) was observed against ampicillin.
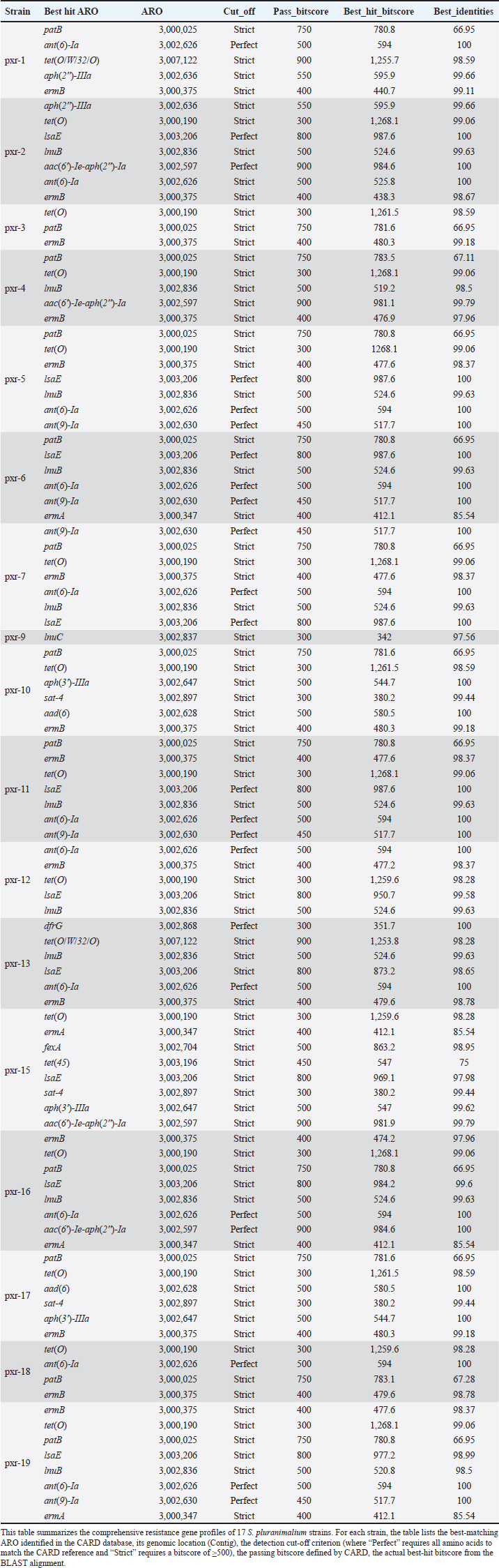
Fig. 1. Antimicrobial resistance profiles of 17 S. pluranimalium isolates. Resistance and susceptibility are indicated by filled and unfilled purple circles, respectively. The left side of the figure displays the bacterial isolate designations and their corresponding accession numbers. The top color bar indicates the antibiotic class for each agent. Table 1. Resistance gene profiles of 17 S. pluranimalium strains detected using the CARD database.
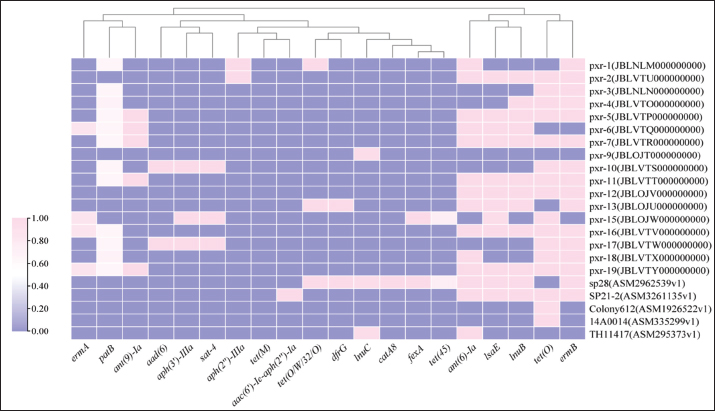
Comparative analysis of the ARGsA total of 17 S. pluranimalium strains were subjected to second-generation sequencing and comparative genomic analysis with 5 additional S. pluranimalium strains, resulting in the identification of 20 distinct antimicrobial resistance genes (Fig. 2). The heatmap visually represents the relative abundance of these genes, with a color gradient from pink (highest abundance) to purple (lowest abundance). Sequencing results revealed significant variations in the carriage rates of these genes. The most prevalent resistance genes were ermB, which confers resistance to macrolides, and tet(O), which is associated with tetracycline resistance, both exhibiting a high carriage rate of 72.73%. These were followed by patB and ant(6)-Ia, involved in β-lactam resistance, with detection rates of 54.55% and 63.6%, respectively. The IsaE gene, which mediates cross-resistance to pleuromutilins, lincosamides, and streptogramin A, also showed a high prevalence (54.55%). Various aminoglycoside resistance genes were identified, including aph(3')-IIIa and sat-4 (both 13.64%) and aph(2'')-Ia and aad(6) (both 9.10%). Notably, the fexA gene, which is responsible for florfenicol resistance, and the dfrG gene, which is associated with trimethoprim resistance, were both detected at a frequency of 9.10%. The hierarchical clustering of isolates based on their ARG profiles revealed a distinct phylogenetic pattern with potential epidemiological significance (Fig. 2). The majority of the porcine-derived isolates from this study (e.g., pxr-1, pxr-2, and pxr-3) formed a primary cluster, which was clearly separate from a subcluster containing other porcine isolates (e.g., pxr-9 and pxr-10) and the publicly available swine-derived strain SP28 from Shandong, China. This clear separation indicates notable differences in the combinatory profiles of AMR genes carried by these distinct clusters. In contrast, the reference strains from non-porcine or international sources—specifically, the environmental/food-associated strains SP21-2 (Sichuan, China) and Colony612 (Thailand), as well as the clinical strain 14A0014 (Switzerland)—consistently formed outgroups, demonstrating a more divergent resistome. This clustering pattern strongly indicates that ARG carriage in S. pluranimalium is not random but is influenced by host and geographical factors. These findings underscore the presence of diverse and complex multidrug resistance mechanisms within this bacterial population.
Fig. 2. Heatmap and hierarchical clustering of AMR genes in S. pluranimalium isolates. The heatmap illustrates the relative abundance of various AMR genes across different samples. Pink indicates the highest relative abundance, whereas purple represents a gradual decrease in abundance. The heatmap displays 22 strains (17 isolates obtained in this study and 5 reference strains), with the rightmost column corresponding to the strain designations and their associated accession numbers for the 22 S. pluranimalium isolates. The dendrograms on the top sides of the heatmap illustrate the hierarchical clustering of isolates and ARGs. DiscussionThis study presents the first systematic characterization of AMR in S. pluranimalium isolates from pigs in Jilin Province, China, revealing a concerning profile of multidrug resistance. All isolates were multidrug-resistant, exhibiting particularly high rates of resistance to sulfonamides (sulfamethoxazole, 100%), macrolides (azithromycin, 94.12%; tylosin tartrate, 70.59%), and tetracyclines (tetracycline, 76.47%). Given these exceptionally high resistance rates, the clinical efficacy of these antimicrobial classes is expected to be severely compromised; thus, they are not recommended for treating infections caused by such resistant strains. This finding is of substantial clinical concern because these antibiotic classes, particularly macrolides and tetracyclines, have been among the first-line or commonly used agents for treating streptococcal infections and respiratory diseases in swine worldwide. This finding is in strong agreement with the global trend of AMR in animal-derived streptococci. For instance, a study on bovine streptococci from North America similarly reported a high prevalence of macrolide and tetracycline resistance, attributed to the extensive use of these antibiotics in livestock production (Tan et al., 2021). This consistency across regions and hosts suggests that resistance to macrolides and tetracyclines may now be a widespread and entrenched feature in animal streptococcal populations, thereby indicating that veterinary clinical practice is urgently needed. However, our results also revealed critical discrepancies and positive findings. Most notably, while resistance to ampicillin was high (88.24%), all isolates remained fully susceptible to penicillin, amoxicillin, and ceftizoxime. This finding suggests a clear and potentially effective therapeutic option for animals infected with multidrug-resistant S. pluranimalium. This result is fully congruent with the global consensus on streptococcal therapy, which upholds β-lactam antibiotics (particularly penicillin and amoxicillin) as first-line agents due to their bactericidal activity and historically low resistance rates among streptococci (Aggarwal et al., 2024). The disparity in resistance between ampicillin and other β-lactams could be due to the action of specific β-lactamases with higher affinity for ampicillin or subtle alterations in penicillin-binding proteins. Nevertheless, the preserved efficacy of penicillin and amoxicillin provides a reliable treatment avenue against this multidrug-resistant pathogen. Comparative genomic analysis based on ARG profiles provided further epidemiological insight, revealing a distinct population structure between our 17 porcine isolates and the five public reference strains. Most of our isolates (e.g., pxr-1, pxr-2, and pxr-3) formed a primary cluster, which was closely related to the swine-derived strain SP28 from Shandong, China. This suggests the potential circulation of a dominant clonal complex or a shared local pool of resistance genes among porcine hosts in geographically proximate regions of China. In stark contrast, strains from environmental/food sources (SP21-2 from Sichuan, China, and Colony612 from Thailand) and a clinical strain from abroad (14A0014 from Switzerland) consistently formed outgroups with divergent resistomes. This clear clustering pattern strongly indicates that the resistome of S. pluranimalium is not random but is significantly shaped by host species and geographical factors, underscoring that our porcine isolates represent a unique population with distinct AMR gene carriage. Within this unique population context, a core novelty of our study lies in the identification of previously unreported combinations of AMR genes in these porcine isolates. We not only report the first co-occurrence of five aminoglycoside resistance genes (aad(6), aph(3')-IIIa, sat-4, aph(2'')-Ia, and patB) in this species but also the simultaneous detection of lnuB and lnuC. More importantly, these genes were found in specific combinatory patterns, such as the frequent co-location of ermB, tet(O), patB, and IsaE, hinting that they might be disseminated as a "resistance gene module" within the population (Wang et al., 2022). This complex genetic repertoire suggests that porcine S. pluranimalium in this region is under intense antibiotic selection pressure and has evolved efficient resistance mechanisms (Elbehiry and Marzouk, 2025). These genes often reside on mobile genetic elements, such as plasmids and transposons, facilitating their dissemination via horizontal gene transfer across bacterial populations (even across genera such as Staphylococcus or Enterococcus). From a One Health perspective, the development of such a complex resistome in food animals constitutes a significant reservoir from which these genes could potentially transfer to human pathogens via direct contact, the food chain, or environmental contamination, thereby compromising our ability to treat bacterial infections in humans. Correlating phenotypes with genotypes revealed a complex picture. A strong correlation was evident between macrolide and tetracycline resistance, with the high prevalence of ermB and tet(O) genes (72.73% each) corresponding well with the observed high-level resistance. However, notable discordances were also apparent. The 100% phenotypic resistance to sulfamethoxazole, in the absence of detected sulfonamide resistance genes (e.g., sul genes), strongly implies the involvement of yet-unidentified resistance mechanisms, such as unknown sul gene variants or altered metabolic pathways (Wang et al., 2023; Tan et al., 2021; Boolchandani et al., 2019). Similarly, the discrepancy between the low detection rate of the phenicol-specific efflux pump gene fexA (9.10%) and the moderate resistance to florfenicol (52.94%) suggests the contribution of alternative mechanisms, such as the upregulation of non-specific efflux pumps or the presence of floR-like genes not annotated in current databases. These inconsistencies serve as a critical reminder that genotype-based predictions have limitations, and phenotypic susceptibility testing remains indispensable for accurately guiding clinical therapy. In conclusion, this study presents the first comprehensive overview of MDR in porcine S. pluranimalium from China. We confirmed high-level resistance to multiple antibiotic classes and uncovered a distinct population structure with geographical and host-specific characteristics, harboring novel and complex resistance gene combinations, through comparative genomics. The potential for these genes to spread via horizontal gene transfer underscores the urgent need for enhanced antibiotic stewardship and AMR surveillance in veterinary sectors (O’Neill, 2016). Fortunately, sustained susceptibility to key β-lactam antibiotics preserves a vital therapeutic window. Future research should focus on elucidating the genetic context and mobility of these ARGs by characterizing the plasmids or integrative conjugative elements that carry them and conducting conjugation experiments to directly confirm their transferability. This will enable a more comprehensive risk assessment of their dissemination potential. AcknowledgmentsWe extend our appreciation to the Jilin Province Science and Technology Innovation Platform Subsidy Project and JiLin Agricultural Science and Technology College staff members. Conflict of interestThe authors declare no conflicts of interest in the work reported in this paper. FundingThis research was funded by the Jilin Province Science and Technology Innovation Platform Subsidy Project (Grant number YDZJ202302CXJD038). Author contributionsMethodology: Hannuo Li.; Software: Xiaorui Pan.; Validation: Nuo Xu. and Xiaorui Pan.; Formal Analysis: Hannuo Li.; Investigation: Hannuo Li. and Xiaorui Pan.; Resources: Guangyan Zhang; Data Curation: Nuo Xu; Visualization: Guangyan Zhang.; Writing—Original Draft Preparation: Hannuo Li.; Writing—Review & Editing: Wanli Sha. Baishuang Yin, Wenlong Dong, Wanli Sha Baishuang Yin and Wenlong Dong; Wanli Sha Project Administration Baishuang Yin and Wenlong Dong; Funding Acquisition: Wenlong Dong. Ethical statementBefore sample collection at pig farms, verbal consent was obtained from the farm authorities following proper notification. All sampling activities were conducted in accordance with animal welfare protocols approved by the Institutional Animal Care and Use Committee of JiLin Agricultural Science and Technology College, with farm operators providing verbal informed consent. This research was approved by the Animal Ethics Committee of JiLin Agricultural Science and Technology College (Approval No. LLSC202301055). Date: 2/01/2023. Data availabilityData from the results of this study are not publicly available due to sensitivity. However, they can be obtained from the corresponding author upon reasonable request. Data are stored in a controlled-access data storage facility at JiLin Agricultural Science and Technology College ReferencesAggarwal, R., Mahajan, P., Pandiya, S., Bajaj, A., Verma, S.K., Yadav, P., Kharat, A.S., Khan, A.U., Dua, M. and Johri, A.K. 2024. Antibiotic resistance: a global crisis, problems, and solutions. Crit. Rev. Microbiol. 50(5), 896–921; doi:10.1016/j.crm.2011.08.021 Ahmed, A., Zaman, G., Gardezi, A., Satti, L.L., NorthN, S. and Haleem, A. 2018. Cerebral abscess caused by novel species: Streptococcus pluranimalium. J. Coll. Physicians. Surg. Pak. 28(9), 181–183; doi:10.1016/j.jcpsp.2008.08.037 Boolchandani, M., D’Souza, A.W. and Dantas, G. 2019. Sequencing-based methods and resources to study antimicrobial resistance. Nature Rev. Genet. 20(6), 356–370. Chen, C., Wu, Y., Li, J., Wang, X., Zeng, Z., Xu, J., Liu, Y., Feng, J., Chen, H., He, Y. and Xia, R. 2023. TBtools-II: a 'one for all, all for one' bioinformatics platform for biological big-data mining. Mol. Plant. 16(10), 1733–1742. Devriese, L.A., Vandamme, P., Collins, M.D., Alvarez, N., Pot, B., Hommez, J., Butaye, P. and Haesebrouck, F. 1999. Streptococcus pluranimalium sp. nov., from cattle and other animals. Int. J. Syst. Evol. Microbiol. 49(3), 1221–1226. Elbehiry, A. and Marzouk, E. 2025. From Farm to Fork: antimicrobial-Resistant Bacterial Pathogens in Livestock Production and the Food Chain. Vet. SciVeterinary. Sci. 12(9), 862. Haenni, M., Lupo, A. and Madec, J.Y. 2018. Recombination between ermB and tet(O) genes in Streptococcus pyogenes and Streptococcus dysgalactiae subsp. equisimilis isolates. J. Antimicrob. Chemotherapy. 73(5), 1243–1246. Kalhoro, D.H., Luo, S., Xie, X., Zhao, Y.B., Lu, C.P. and Liu, Y.J. 2015. Streptococcus pluranimalium isolated from a canine respiratory case: identification and experimental infection in mice. Am. J. Anim. Vet. Sci. 10(4), 256–262; doi:10.1016/j.ajavisci.2016.01.002 Moreno, C., Romero, J. and Espejo, R.T. 2002. Polymorphism in repeated 16S rRNA genes is a common property of type strains and environmental isolates of the genus Vibrio. Microbiology 148(4), 1233–1239. | ||
| How to Cite this Article |
| Pubmed Style Li H, Pan X, Xu N, Zhang G, Sha W, Yin B, Dong W. Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, China. Open Vet. J.. 2026; 16(2): 1213-1221. doi:10.5455/OVJ.2026.v16.i2.39 Web Style Li H, Pan X, Xu N, Zhang G, Sha W, Yin B, Dong W. Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, China. https://www.openveterinaryjournal.com/?mno=284393 [Access: February 27, 2026]. doi:10.5455/OVJ.2026.v16.i2.39 AMA (American Medical Association) Style Li H, Pan X, Xu N, Zhang G, Sha W, Yin B, Dong W. Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, China. Open Vet. J.. 2026; 16(2): 1213-1221. doi:10.5455/OVJ.2026.v16.i2.39 Vancouver/ICMJE Style Li H, Pan X, Xu N, Zhang G, Sha W, Yin B, Dong W. Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, China. Open Vet. J.. (2026), [cited February 27, 2026]; 16(2): 1213-1221. doi:10.5455/OVJ.2026.v16.i2.39 Harvard Style Li, H., Pan, . X., Xu, . N., Zhang, . G., Sha, . W., Yin, . B. & Dong, . W. (2026) Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, China. Open Vet. J., 16 (2), 1213-1221. doi:10.5455/OVJ.2026.v16.i2.39 Turabian Style Li, Hannuo, Xiaorui Pan, Nuo Xu, Guangyan Zhang, Wanli Sha, Baishuang Yin, and Wenlong Dong. 2026. Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, China. Open Veterinary Journal, 16 (2), 1213-1221. doi:10.5455/OVJ.2026.v16.i2.39 Chicago Style Li, Hannuo, Xiaorui Pan, Nuo Xu, Guangyan Zhang, Wanli Sha, Baishuang Yin, and Wenlong Dong. "Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, China." Open Veterinary Journal 16 (2026), 1213-1221. doi:10.5455/OVJ.2026.v16.i2.39 MLA (The Modern Language Association) Style Li, Hannuo, Xiaorui Pan, Nuo Xu, Guangyan Zhang, Wanli Sha, Baishuang Yin, and Wenlong Dong. "Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, China." Open Veterinary Journal 16.2 (2026), 1213-1221. Print. doi:10.5455/OVJ.2026.v16.i2.39 APA (American Psychological Association) Style Li, H., Pan, . X., Xu, . N., Zhang, . G., Sha, . W., Yin, . B. & Dong, . W. (2026) Antimicrobial resistance profiles and genomic characterization of Streptococcus pluranimalium isolates from pigs in Jilin Province, China. Open Veterinary Journal, 16 (2), 1213-1221. doi:10.5455/OVJ.2026.v16.i2.39 |

