




| Review Article | ||
Open Vet. J.. 2025; 15(12): 6190-6214 Open Veterinary Journal, (2025), Vol. 15(12): 6190-6214 Review Article Hepatic stellate cells in liver fibrosis: From biology to pathologyHicham Esselmani1*, Youssef Nadir1, Rahma Ennadi1, Nadia Zouhairi1, Omar El Hiba2, Mohammed Amine Lkousse3, Mustapha Najimi4,5,# and Mohamed Merzouki1#1Faculty of Science and Technology, Biological Engineering Laboratory, Sultan Moulay Slimane University, Beni Mellal, Morocco 2Biotechnologies and Health Laboratory, Neurosciences and Toxicology Team, Faculty of Sciences, Chouaib Doukkali University, El Jadida, Morocco 3Faculty of Medicine and Pharmacy, Sultan Moulay Slimane University, Beni Mellal, Morocco 4Laboratory of Pediatric Hepatology and Cell Therapy, Institute of Experimental and Clinical Research, Brussels, Belgium Private University of Marrakech, Marrakech, Morocco *Corresponding Author: Hicham Esselmani. Faculty of Science and Technology, Biological Engineering Laboratory, Sultan Moulay Slimane University, Beni Mellal, Morocco. Email: esselmani1 [at] yahoo.fr Submitted: 12/08/2025 Revised: 22/10/2025 Accepted: 02/11/2025 Published: 31/12/2025 © 2025 Open Veterinary Journal
AbstractHepatic stellate cells (HSCs) are pivotal in the development of liver fibrosis, a serious condition characterized by excessive extracellular matrix (ECM) accumulation and compromised liver function. HSCs remain dormant in a healthy liver, effectively storing vitamin A and ensuring ECM stability. However, when faced with injuries from viral hepatitis, alcohol abuse, or nonalcoholic steatohepatitis, these cells are activated and transform into proliferative, ECM-producing myofibroblasts. This review provides a comprehensive overview of the biology of HSCs and their transition from a quiescent to an activated state. The key signaling pathways (e.g., transforming growth factor-beta, platelet-derived growth factor, Wnt/β-catenin) and cellular interactions that drive hepatocyte stem cell (HSC) activation are examined, with a special emphasis on emerging themes such as metabolic reprogramming and epigenetic control. Furthermore, the landscape of antifibrotic therapies targeting HSCs, from pathway inhibitors and epigenetic drugs to ribonucleic acid based strategies, is critically evaluated, and their translational potential and challenges are discussed. By integrating foundational knowledge with recent advances—including insights from single-cell technologies revealing HSC heterogeneity—this review aims to offer a timely and critical perspective on the pathobiology of HSCs and the evolving strategies to combat liver fibrosis. Keywords: Hepatic stellate cells; Extracellular matrix; Liver fibrosis; Fibrosis therapeutics. IntroductionThe liver is an essential organ with a remarkable ability to regenerate and adapt. Central to its physiology and pathology are hepatic stellate cells (HSCs)—specialized perisinusoidal cells that maintain liver homeostasis and drive fibrogenesis. First described in the late 19th century for their star-like morphology, HSCs are now recognized as multifunctional cells that regulate extracellular matrix (ECM) turnover, vitamin A storage, sinusoidal blood flow, and immune signaling (Higashi et al., 2017; Tsuchida and Friedman, 2017). In their quiescent state, HSCs contain cytoplasmic lipid droplets rich in retinyl esters, which account for approximately 80% of the total vitamin A stores in the body (Senoo et al., 2010). This phenotype, resembling adipocytes, is maintained by adipogenic transcription factors such as peroxisome proliferator-activated receptor gamma (PPARγ) and C/EBPs, and by crosstalk with hepatocytes, Kupffer cells, and sinusoidal endothelial cells. HSCs become activated upon liver injury, losing their vitamin A content and differentiating into proliferative, contractile, and fibrogenic myofibroblast-like cells that produce ECM components and drive fibrosis (Bataller and Brenner, 2005; Kisseleva and Brenner, 2021). Hepatocyte stem cell (HSC) activation is driven by a myriad of signaling pathways and mediators, including transforming growth factor-beta (TGF-β), platelet-derived growth factor (PDGF), and reactive oxygen species (ROS) (Inagaki and Okazaki, 2007; Lee and Friedman, 2011). These factors promote ECM deposition and alter the metabolic and functional properties of HSCs, shifting them from a quiescent, vitamin A-storing state to an activated, fibrogenic myofibroblast-like phenotype (Hernandez-Gea and Friedman, 2011). This transition is accompanied by changes in gene expression, cytoskeletal organization, and cell-cell interactions, making HSCs a central focus of liver pathology research (Mederacke et al., 2013). HSCs are involved in liver diseases such as chronic hepatitis, non-alcoholic steatohepatitis, alcoholic liver disease, and hepatocellular carcinoma (Coulon et al., 2011; Parola and Pinzani, 2019). Persistent HSC activation causes scar buildup, disrupting liver structure and function, during chronic injury. In addition, activated HSCs influence the tumor environment by releasing growth factors and cytokines that promote cancer growth, blood vessel formation, and immune evasion (Mazzocca et al., 2011; Coulouarn and Clément, 2014). Although we have made substantial progress in understanding HSC biology, many aspects of their origin, regulation, and therapeutic targeting remain poorly understood. The embryonic origin of HSCs remains debatable, with evidence suggesting that they may derive from the mesoderm, neural crest, or even bone marrow-derived cells (Cassiman et al., 2002; Asahina et al., 2011). Emerging insights from single-cell ribonucleic acid (RNA) reveal substantial heterogeneity within HSCs, with distinct subpopulations exhibiting diverse health and disease responses (Alexandra et al., 2024; Du et al., 2024). These subpopulations are linked to disease causes and fibrosis stages, paving the way for personalized anti-fibrotic therapies targeting specific harmful subsets (Geng and Schwabe, 2025). Although multiple strategies aim to inhibit HSC activation or promote quiescence, translating these strategies into effective therapies remains challenging (Schuppan and Kim, 2013; Trautwein et al., 2015). Our work uniquely combines foundational HSC biology with advances in single-cell technologies, cellular heterogeneity, and metabolic reprogramming, while critically assessing challenges in translating these therapies. This review aims to provide a comprehensive and timely critical analysis of the biology of HSCs, covering their developmental origins, physiological functions, and role in liver disease. We examine and place a special emphasis on the mechanisms underlying HSC activation, their interactions with other liver cell types, and their contributions to liver fibrosis and cancer. Finally, we critically assessed current and emerging therapeutic strategies targeting HSCs, highlighting the potential for new treatments to prevent or reverse liver disease. Biology of hepatic stellate cellsOrigin and developmentHepatic stellate cells become recognizable during the latter half of the third month (days 75–90) of human development (Semba et al., 1983). Stellate cells also appear in the second half of the third month of embryonic development. They are located in the Disse space and contain 1–2 lipid droplets. In a 5-month-old human fetus, stellate cells have small lipid droplets (Wake, 1980). At birth, stellate cells have not yet reached their final shape and size (Wake et al., 1991). In a 1-day-old neonatal rat, stellate cells appear somewhat rounded, with thin and sparse processes. At 2–3 weeks post-birth, these processes extend and develop into perforated membranous appendages. At 5 weeks, the stellate cells mature into their final dendritic shape, featuring elongated perisinusoidal and thicker intersinusoidal extensions (Fig. 1) (Wake et al., 1991).
Fig. 1. Development of HSCs. Single-cell proteomic trajectory analysis has conclusively shown that cells destined to become HSCs express markers linked to fetal liver mesothelium during differentiation, confirming their developmental lineage (Lua and Asahina, 2016; Du et al., 2024). In the adult liver, hepatic stellate cells express mesenchymal markers, such as vimentin, desmin, and alpha-smooth muscle actin (α-SMA), or neural/neuroectodermal markers, such as glial fibrillary acidic protein (GFAP), neural cell adhesion molecule, and synaptophysin (Schmitt-Gräff et al., 1993; Neubauer et al., 1996; Cassiman et al., 1999; Geerts, 2001). Based on these characteristic phenotypes, the embryonic origin of hepatic stellate cells is believed to be either the septum transversum mesenchyme or the neural crest (Friedman, 2000; Geerts, 2001). Cassiman et al. found that hepatic stellate cells do not originate from the neural crest in transgenic mice expressing yellow fluorescent protein in all neural crest cells and their derivatives. This suggests that they may derive from the septum transversum mesenchyme, endoderm, or mesothelium (Cassiman et al., 2006). Lineage-tracing studies have provided crucial evidence for the mesothelial origin of HSCs. Kendall et al. (2019) demonstrated that a significant population of quiescent HSCs in the adult liver originates from the embryonic Wilms' tumor homolog (WT1)-positive mesothelium using advanced techniques in mice. The current consensus, supported by a growing body of evidence, strongly points to a mesodermal origin, specifically from the mesothelium of the septum transversum, which gives rise to most liver mesenchymal cells and mesenchymal cells seem to be trapped between growing hepatoblasts and become HSCs. Conditional cell-lineage tracing in mice revealed that Wilms tumor homolog WT1+ cells in the septum trans-versum mesenchyme give rise to mesothelial cells on the liver surface and HSCs in the liver during liver development (Kendall et al., 2019; Du et al., 2024). Quiescent stellate cells express various neural markers, including GFAP, nestin, Nerve growth factor, brain-derived growth factor, NT-3, and NT-4/5, as well as p75, tropomyosin receptor kinase B and C. They also produce the cell adhesion protein neural cell adhesion molecule, the prion protein cellular prion protein, and synaptophysin (Geerts, 2001). In addition, their dendritic morphology resembles that of astrocytes rather than the spindle shape typical of quiescent skin fibroblasts. These observations suggest that stellate cells may originate from neuroectodermal tissue (Niki et al., 1999; Friedman, 2000). Embryonic neural tissues consist of multipotent cells that differentiate into different sublineages based on growth factors. These cells become glial cells when exposed to neuregulin isoform I; under BMP-2 influence, they develop into neurons; and when incubated with TGF-β, they differentiate into myofibroblasts or smooth muscle cells (Hagedorn et al., 1999; Morrison et al., 1999). The origin of stellate cells has been a subject of debate. They may originate from either the endoderm or the transversal septum among various tissues (Asahina et al., 2011; Chan et al., 2013). Evidence supporting the septum transversum origin includes the expression of the mesodermal transcription factor Foxf1 in HSCs, which is specifically found in the septum transversum during liver development (Chan et al., 2013). Additionally, some researchers propose that HSCs and hepatoblasts share a common endodermal origin, based on the shared expression of specific cytokeratin proteins in both cell types (Kiassov et al., 1995; Levy et al., 1999). Crude bone marrow (BM) cells can engraft into the hepatic stellate cells of lethally irradiated mice (Baba et al., 2004; Russo et al., 2006). Because adult BM contains both hematopoietic and mesenchymal stem cells, the specific cell type responsible remains uncertain. Earlier studies suggested that glomerular mesangial cells in the kidney and perivascular pericyte-like cells in the brain are derived from hematopoietic stem cells in mice that received single hematopoietic stem cell transplants (Masuya et al., 2003; Hess et al., 2004). Interestingly, both cell types are considered to belong to the myofibroblast family. Fibroblasts and myofibroblasts in many organs and tissues originate from hematopoietic stem cells (Ogawa et al., 2006). In a mouse model, Miyata et al. (2008) reported that hematopoietic stem cells can give rise to hepatic stellate cells during liver fibrosis (Miyata et al., 2008). However, the exact process by which hematopoietic stem cells differentiate into hepatic stellate cells remains unclear. The mesothelium originating from the mesoderm is a key source of HSCs and other mesenchymal cells in the liver, such as portal fibroblasts. In the adult liver, bone marrow-derived cells are also believed to contribute to HSCs or myofibroblasts, although this contribution is likely minimal (Baba et al., 2004; Higashiyama et al., 2009). Hematopoiesis occurs in the liver during embryonic development, with hematopoietic stem cells migrating to the bone marrow before birth. Consequently, the primary source of HSCs during embryogenesis is probably the mesothelium derived from the septum transversum mesenchyme, rather than bone marrow cells. Structure and function of theHSCs, also called Ito cells, vitamin A-storing cells, lipocytes, or fat-storing cells, have several key functions: storing and maintaining retinoids; remodeling the ECM by producing ECM components and matrix metalloproteinases (MMPs); generating growth factors and cytokines; and adjusting the sinusoidal lumen through contraction and dilation in response to endothelin, angiotensin, or prostaglandins (Sato et al., 2003). In a healthy liver, stellate cells feature spindle-shaped bodies with oval or elongated nuclei. Their perikaryons are situated in the recesses between the adjacent parenchymal cells. Ultrastructurally, they consist of a moderately developed rough endoplasmic reticulum (rER), a small Golgi apparatus close to the nucleus (Enzan et al., 1999), and prominent dendritic processes extending into the cytoplasm (Wake 1999). The subendothelial processes form around sinusoids located between ECs and hepatocytes. Each process displays many thorn-like microprojections (Wake 1995). Retinoids (vitamin A and its derivatives) are known to regulate various cellular processes, such as cell growth, differentiation, morphogenesis, and tumorigenesis (Blomhoff, 1994; Chawla et al., 2001). Quiescent stellate cells store 80% of the body’s retinoids, mainly as retinyl palmitate in their cytoplasmic lipid droplets, and help regulate the transport and storage of retinoids (Blomhoff et al., 1990; Senoo, 2004). Retinol and retinol-binding protein (RBP) complexes circulate in the bloodstream. It binds specifically to receptors on the surface of stellate cells and is then transported into endosomes via pits and vesicles. Within the endosomes, retinol can follow three pathways: (1) binding to cellular retinol-binding protein (CRBP), followed by esterification and storage in lipid droplets; (2) oxidation to retinoic acid, which then binds to CRBP or nuclear receptors (Retinoic acid receptor / retinoid X receptor) to regulate gene expression; (3) transport back to the endoplasmic reticulum for re-binding with RBP and secretion from the cell (Senoo, 2004). In a typical liver, the space separating parenchymal cells from HSCs and endothelial cell complexes contains fibrillar ECM components, such as type I and type III collagen, along with basement membrane elements. However, these usually do not display an electron-dense structure typical of basement membranes in other tissues. The main component produced is type I collagen, with minor amounts of type III and type IV collagen (Senoo et al., 1984). Hepatic stellate cells lose retinoids, proliferate actively, and produce and secrete large amounts of ECM components such as collagen, proteoglycans, and glycoproteins in pathological conditions like liver cirrhosis. The typical ECM components, including parenchymal cells, endothelial cells, and HSCs, are crucial for maintaining the differentiated functions and structure of resident liver cells. During liver fibrosis, the composition of the hepatic ECM changes both quantitatively and qualitatively. The total levels of collagenous and non-collagenous ECM components increase, and the ECM between endothelial cells and HSCs shifts from typical basement membrane-like matrices to fibrous, collagen-rich matrices (Friedman, 2000). HSCs produce various types of MMPs. These enzymes, including MMP-1, MMP-2, MMP-3, metalloproteinase 9, and MMP-11, are essential for ECM remodeling during liver injury and regeneration. MMPs are expressed in both HSCs and hepatic parenchymal cells (Geisler et al., 1997), whereas tissue inhibitors of metalloproteinases 1 and 2 (TIMPs-1 and TIMP-2) are expressed exclusively in HSCs (Herbst et al., 1997). Transforming growth factor β1 (TGF-β1) and PDGF are well-characterized fibrogenic and proliferative cytokines, respectively, for HSCs. TGF-β1 is elevated in experimental and human hepatic fibrosis and induces ECM production by HSCs (Friedman, 2000). Although various sources of this cytokine exist, autocrine expression in HSCs is considered the most significant (Gressner, 1995). Role of HSCs in liver metabolism regulationQuiescent hepatic stellate cells (qHSCs) are characterized by cytoplasmic lipid droplets that store large amounts of vitamin A metabolites or retinoids. This distinctive feature led to their identification by Karl Wilhelm von Kupffer, who used gold chloride staining to highlight their presence (Kupffer, 1876). More than 95% of these retinoids are stored as REs, mainly in the form of long-chain acyl moieties such as retinyl-palmitate, retinyl-stearate, retinyl-oleate, and retinyl-linoleate, which constitute about 30%–50% of the lipid droplet content in HSCs (Senoo et al., 2010). These lipid droplets contain triglycerides, cholesterol, cholesterol esters, phospholipids, and free fatty acids (Moriwaki et al., 1988). Retinol esterification, which is the process of storing retinol in lipid droplets, is mainly controlled by the enzyme lecithin: retinol acyltransferase (Ajat et al., 2017). Although acyl-CoA: retinol acyltransferase also has a minor role (O’Byrne et al., 2005), Lecithin:retinol acyltransferase (LRAT) knockout (LRAT−/−) mice cannot store HSC lipid droplets or hepatic retinyl esters (O’Byrne et al., 2005). Retinol helps maintain the quiescent state of HSCs through interactions with retinoic acid receptor β and retinoid X receptor α (Wang et al., 2002). Vitamin A treatment can prevent HSC activation in culture and maintain the expression of markers linked to the quiet state, such as GFAP and PPAR-γ, while also lowering the expression of activation markers like αSMA, Col1a1, and HSP-47 (Yoneda et al., 2016). When activated, HSCs decrease LRAT expression, which reduces their ability to form and store retinyl esters (Shmarakov et al., 2019), leading to the loss of retinoids essential for maintaining their quiescent state. This loss may contribute to systemic vitamin A deficiency in patients with liver diseases. In addition to their role in retinoid storage, HSCs also regulate lipid metabolism through the expression of PPARγ and sterol regulatory-element-binding protein-1c (SREBP-1c). These proteins regulate fatty acid storage and metabolism in quiescent cells. Both PPARγ and SREBP-1c are highly expressed in qHSCs but are significantly downregulated upon activation (Tsukamoto, 2005). Increasing the expression or activity of PPARγ and SREBP-1c reverses HSC activation, suggesting that maintaining the adipogenic phenotype preserves quiescence (Tsukamoto, 2005). Conversely, knocking down PPARγ in cultured human HSCs significantly increases activation (Tao et al., 2020). Interestingly, despite the complete loss of lipid droplets in LRAT−/− HSCs, these cells display maintained expression of PPARγ and SREBP-1c, perhaps explaining their preserved quiescent phenotype (O’Byrne et al., 2005). HSCs also express the “adipokines” leptin and adiponectin, and adiponectin signaling is antifibrotic (Kamada et al., 2003). Upon activation, HSCs show increased expression of PPARβ, a regulator of fatty acid oxidation enzymes. In contrast, the expression of peroxisome proliferator-activated receptor alpha (She et al., 2005), which is involved in fat oxidation, is surprisingly reduced in activated HSCs. This altered expression of lipid metabolism regulators highlights the dynamic role of HSCs in liver metabolism, where their function shifts from fat storage to fibrosis and tissue remodeling upon activation (Miyahara et al., 2000). Several enzymes, such as adipose triglyceride lipase, patatin-like phospholipase domain-containing protein 3, hormone-sensitive lipase, and carboxyl ester lipase, are crucial for releasing retinyl esters from lipid droplets (Taschler et al., 2015). Blocking fatty acid oxidation can prevent HSC activation (Hernández–Gea et al., 2012), indicating that fatty acid supply from lipid droplets might be essential for this process. Additionally, HSC activation leads to increased glucose and amino acid breakdown (Trivedi et al., 2021). Upon activation, qHSCs undergo a fundamental metabolic reprogramming, shifting to aerobic glycolysis. This "glycolytic switch," analogous to the Warburg effect observed in cancer cells, is characterized by increased glucose uptake and its conversion to lactate, irrespective of oxygen availability. This adaptation provides both the rapid adenosine triphosphate (ATP) generation required for their proliferation and the biosynthetic precursors essential for the massive production of ECM, thereby supporting their fibrogenic phenotype (Roh et al., 2025). This enhanced glucose utilization does increase mitochondrial oxidation, but it more notably boosts glycolytic flux and lactate production, which is believed to directly contribute to activation (Chen et al., 2012). Conversely, reducing mitochondrial pyruvate metabolism, which would theoretically raise intracellular lactate levels, has been suggested to decrease HSC activation (McCommis et al., 2017). In response to elevated extracellular glucose levels or purinergic signaling, activated HSCs show higher rates of glucose utilization, improved glucose transport capabilities, and increased glycolytic activity (Yin et al., 2023). This phenomenon is attributed to the upregulation of mRNA expression of glucose transporters, including GLUT1, GLUT2, and GLUT4 (Yan, 2017). Notably, liver cancer cells, which mainly generate ATP through anaerobic metabolism, also demonstrate significant overexpression of GLUT1, which is a rate-limiting step in ATP synthesis (Du et al., 2022). Additionally, activated HSCs exhibit increased mRNA expression of rate-limiting glycolytic enzymes, including hexokinase-2, pyruvate kinase M2, and fructose-2,6-bisphosphatase-3 (Du et al., 2022). Increased glycolysis in HSCs during cultivation leads to the depletion of key central carbon metabolites from the citric acid cycle. HSC activation is marked by high levels of pyruvate dehydrogenase kinase 3, which blocks the transformation of pyruvate into acetyl-CoA, instead leading to increased lactate production. Lactate is essential for HSC activation and myofibroblast phenotype survival. Although the lactate export pump, monocarboxylate transporter 4, is upregulated, the lactate levels inside activated HSCs stay high. Notably, inhibiting lactate intracellular accumulation reduces cell proliferation, lowers the expression of genes linked to the myofibroblast profile, and enhances lipid storage and lipogenic gene activity (Karthikeyan et al., 2016). Amino acids, especially glutamine, play a crucial role in fueling the activation and fibrogenic activity of HSCs. Upon activation, HSCs enhance glutaminolysis—the breakdown of glutamine to α-ketoglutarate—which serves as a major anaplerotic substrate for the tricarboxylic acid cycle. This helps meet the high bioenergetic and biosynthetic demands of proliferating activated HSCs (aHSCs), especially when glucose is preferentially diverted toward glycolysis (Roh et al., 2025). Interaction between human stem cells and other liver cellsKupffer cells, situated within the perisinusoidal space, have a unique ability to influence the behavior of stellate cells. They release various regulatory substances, including superoxide, nitric oxide (NO), and cytokines such as tumor necrosis factor (TNF), interleukin-6 (IL-6), transforming growth factor-α, and TGF-β. Kupffer cells stimulate the proliferation of stellate cells (Gressner and Zerbe, 1987; Gressner and Haarmann, 1988) and ECM synthesis (Friedman and Arthur, 1989). Proliferation has been attributed to Kupffer cell–derived TGF-β and to a second factor that induces the expression of the PDGF receptor on stellate cells (Friedman and Arthur, 1989). TGF-β from Kupffer cells markedly stimulates stellate cell ECM (Gressner et al., 1993; Gressner, 1995). TNF inhibits collagen synthesis and gene expression (Knittel et al., 1997; Hernandez et al., 2000). Quiescent stellate cells are said to respond more strongly to TGF-β than fully activated ones because they downregulate TGF receptors as they become myofibroblasts (Dooley et al., 2000). Conversely, the response to TNF might be the opposite; some researchers suggest that quiescent stellate cells do not react to TNF even though they have the surface receptors for this cytokine (Hellerbrand et al., 1998). TNF, IL-1, and in some instances lipopolysaccharide (LPS) and interferon (IFN-γ) induce stellate cells to secrete leukocyte chemoattractants and express leukocyte adhesion markers. In these respects, stellate cells can exert reciprocal effects on leukocytes. Kupffer cells are thought to play a significant role in eliciting inflammatory responses in stellate cells because they are prominent sources of TNF and interleukin (IL)-1. They produce both anti-inflammatory and pro-inflammatory cytokines, including IL-10 (Wang et al., 1998). Interestingly, IL-10 seems to inhibit fibrogenesis in stellate cells by reducing collagen production and increasing collagenase secretion (Wang et al., 1998). Additionally, Kupffer cells affect stellate cells by releasing matrix (MMP-9; gelatinase B) (Winwood, 1995), an enzyme capable of activating latent TGF-β, which then promotes collagen synthesis in stellate cells (Yu and Stamenkovic, 2000). In addition, Kupffer cells produce eicosanoids, including prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), and thromboxanes (Peters et al., 1990; Kawada et al., 1992a,b). Eicosanoids regulate stellate cell contractility, with thromboxanes generally promoting contraction and PGE2 encouraging relaxation (Kawada et al., 1992a,b). By upregulating cyclooxygenase-2 and prostaglandin E synthase (Dieter et al., 2000), TNF triggers Kupffer cells to produce PGE2 and thromboxanes, and to a lesser extent PGD2. The impact on PGE2 appears to predominate. Neutrophils are a significant source of ROS, which directly stimulates stellate cell collagen production. Their role as a stimulator of stellate cells was demonstrated in a coculture experiment where cells activated by N-formyl-methionyl-leucyl-phenylalanine were placed in direct contact with stellate cells (Casini, 1997). These activated neutrophils tripled collagen synthesis in stellate cells compared to controls. Superoxide was identified as the primary mediator of this effect. Activated neutrophils also produced NO, which lessened but did not eliminate the impact of superoxide on collagen expression. Lymphocytes also influence stellate function by releasing cytokines. They produce TNF and TGF-β, which can affect stellate cells similarly to Kupffer cells. However, lymphocytes have a broad cytokine profile (Maher, 1999), and many other compounds from these cells can affect stellate cell activity. The cytokines most studied in stellate cells from lymphocytes are IL-4 and IFN-γ. IL-1 and IL-4 promote fibrosis in culture (Tiggelman et al., 1995), while IFN-γ inhibits stellate cell activation (Rockey and Chung, 1994). The fibrogenic effects of IL-4 and the antifibrotic effects of IFN-γ have been confirmed in animal models of fibrosis (Rockey and Chung, 1994). Stellate cells also influence inflammatory cell behavior. Over the last 20 years, their role in immune regulation has become more recognized. The first clues appeared in 1992 when Pinzani et al. (1992) reported that stellate cells produce M-CSF upon stimulation with PDGF or fibroblast growth factor (Pinzani et al., 1992). M-CSF is crucial for maintaining monocyte/macrophage differentiation; therefore, it was thought to help regulate Kupffer cell homeostasis during liver injury. Later studies have shown that stellate cells also produce platelet-activating factor, a lipid chemoattractant (Pinzani et al., 1994). Subsequently, research has shifted to the production of other inflammatory mediators, especially chemokines, by stellate cells. These produce several chemokines, such as MCP-1, MIP-2, and CINC, a rat analog of IL-8 (Marra et al., 1993). These belong to two chemokine families: CC (MCP-1) and CXC (MIP-2 and CINC). Both chemokines attract leukocytes but target different cell types. CC chemokines mainly recruit mononuclear cells, whereas CXC chemokines recruit neutrophils (Baggiolini, 1998). Stellate cells spontaneously start producing chemokines during activation in culture (Marra et al., 1993). Activation of hepatic stellate cellsMechanisms of HSC activationUpon liver injury, qHSCs are activated and transformed into (aHSCs; myofibroblasts). This transition is characterized by loss of lipid droplets and acquisition of a contractile, proliferative, and fibrogenic phenotype, accompanied by notable changes in gene expression (Figure 2) (Liu et al., 2020).
Fig. 2. Changes in microRNA (miRNA) expression during HSC activation. Upon hepatocyte injury, quiescent HSCs (Q-HSCs) become activated HSCs (A-HSCs), losing their vitamin A droplets and developing a myofibroblastic phenotype. During this activation process, several miRNAs are differentially expressed. miRNAs that promote HSC activation are upregulated (red arrow), whereas miRNAs that inhibit HSC activation are downregulated (blue arrow). Excess lipid and cholesterol accumulation in hepatocytes can cause lipotoxicity by generating free radicals, such as ROS. This process leads to oxidative stress, damages cellular metabolism and membrane integrity, and impairs organelle function, including mitochondrial dysfunction and endoplasmic reticulum (ER) stress. In addition, it stimulates the release of proinflammatory cytokines (Ibrahim et al., 2018). Hepatic cholesterol buildup can directly activate hepatic stellate cells (HSCs) by stimulating toll-like receptor 4 signaling or indirectly through Kupffer cells that uptake cholesterol and then activate HSCs by releasing interleukin IL-1β, tumor necrosis factor (TNF), and TGF-β (Musso et al., 2013). The inflammatory response caused by liver injury causes circulating monocytes to migrate to the liver. Together with resident Kupffer cells, these monocytes contribute to HSC activation and fibrosis by producing cytokines such as TGF-β, PDGF, TNF, interleukins, and chemokines (Tsuchida and Friedman, 2017). TNF and IL-1β promote the survival of aHSCs by triggering the NFκB pathway (Pradere et al., 2013). Additionally, IL-1β enhances fibrosis by increasing tissue inhibitors of metalloproteinase 1 (encoded by Timp1) and decreasing bone morphogenetic proteins as well as activin membrane-bound inhibitors. These inhibitors normally function as pseudoreceptors that antagonize TGF-β signaling in HSCs. Hepatocytes change their gene expression and secretions following injury, affecting HSC activation. Damage-Associated Molecular Patterns (DAMPs) released by damaged hepatocytes can directly or indirectly trigger HSC activation. NLRP3, a crucial part of inflammasomes and a downstream target of DAMPs, plays a significant role. Mice with a permanently active NLRP3 mutant develop severe liver inflammation marked by pyroptotic hepatocyte death and HSC activation (Wree et al., 2014). Liver sinusoidal endothelial cells (LSECs) maintain HSCs in a quiescent state in a healthy liver through the release of paracrine factors. Heparin-binding EGF-like growth factor and NO collectively suppress HSC activation (Maretti-Mira et al., 2019; Kisseleva and Brenner, 2021). Normal LSECs are fully differentiated and highly endocytic with fenestrae. These cells lose their fenestration and undergo capillarization before fibrosis develops, a process caused by incomplete differentiation of bone marrow-derived LSECs recruited to the injured liver. These recruited cells are permissive for HSC activation (Xie et al., 2012; Maretti-Mira et al., 2019). Macrophages play dual roles in promoting and resolving liver fibrosis. Activated macrophages are traditionally classified into M1 and M2 types. The M1 phenotype is characterized by high levels of proinflammatory cytokines, increased production of reactive oxygen and nitrogen species, and promotion of Th1 responses (Sica et al., 2014). Enteric dysbiosis can lead to liver pathology through HSC activation. Intestinal imbalance causes the release of pathogen-associated molecular patterns, increasing exposure and triggering HSC activation via toll-like receptors (TLRs) (Zheng et al., 2018). In mice with CCl4 and bile duct ligation, a high-fat diet boosts endotoxin and lipopolysaccharide production by intestinal Gram-negative bacteria, which enhances bacterial translocation and promotes faster fibrous tissue formation by stimulating HSC activation (De Minicis et al., 2014). HSCs activate and develop a myofibroblastic phenotype, characterized by changes in gene expression, following hepatic injury. MicroRNAs (miRNAs) are small noncoding RNA molecules, 19–24 nucleotides long, that are evolutionarily conserved and encoded in the genome. Regulate gene expression by destabilizing mRNA and inhibiting mRNA translation. Specific microRNAs play a key role in HSC activation (Huang et al., 2014; Coll et al., 2015) (Fig. 2). The human genome encodes approximately 1000 miRNAs, which may be expressed ubiquitously or in a tissue- or cell-type specific manner. Each miRNA is likely to have a wide range of potential targets, highlighting their importance in gene regulation. Each miRNA can inhibit the gene expression of several targeted transcripts (Bartel and Chen, 2004; Lee, 2013). miRNAs are recognized as key players in many human diseases, including liver fibrosis and human stem cell transformation. The miR-29 family is one of the earliest and most well-studied miRNA families involved in liver fibrosis and HSC activation (Lambrecht et al., 2015). Multiple microRNAs regulate key fibrogenic signaling pathways in HSCs, including TGF-β/Smad, Wnt/β-catenin, and Hedgehog pathways (Ezhilarasan, 2020). For example, miR-214 is notably increased during HSC activation. It enhances the accumulation of ECM by suppressing the homolog of suppressor-of-fused, which negatively regulates the Hedgehog pathway in LX-2 cells (Ma et al., 2018). miR-214 should be viewed as a key therapeutic target because both genetic and therapeutic silencing approaches have greatly inhibited renal fibrosis (Denby et al., 2014). Additionally, miR-125b promotes HSC activation and fibrogenesis by upregulating RhoA signaling and can serve as an A-HSC-specific fibrosis marker (You et al., 2018). Moreover, epigenetic mechanisms such as DNA methylation may be involved in HSC transformation, with Mann et al. (2007) demonstrating the critical role of DNA methylation in mouse HSC activation. Their findings show that experimental suppression of DNA methylation using 5ʹ-azadeoxycytidine (5ʹ-AzadC) effectively inhibits activation (Mann et al., 2007). Another study by Mann et al. (2010) emphasized the importance of the well-known DNA-binding protein MeCP2, showing that its genetic suppression inhibits HSC activation and protects against liver fibrosis in vivo (Mann et al., 2010). Contemporary research has identified several long non-coding RNAs as key regulators of the cellular mechanisms underlying HSC activation in liver fibrosis, specifically emphasizing the important roles of NEAT1 and H19 in modulating critical signaling pathways and epigenetic modifications during this process. NEAT1 promotes HSC activation through multiple miRNA-mediated mechanisms. It drives fibrogenic signaling by inhibiting miR-139-5p to upregulate the β-catenin/SOX9/TGF-β1 axis and suppressing miR-122, which increases KLF6 expression. Additionally, NEAT1 targets miR-148-3p/miR-22-3p to raise cytohesin 3 levels, represses miR-29b to enhance autophagy via Atg9, and downregulates miR-506 to activate the Hedgehog pathway through GLI3 (Wang et al., 2021; Gao and Mao, 2024). H19 acts as a competing endogenous RNA that worsens liver fibrosis. It binds miR-148a, leading to the overexpression of ubiquitin-specific protease 4 and the continuous activation of the TGF-β pathway by stabilizing Smad4 and TGF-βRI. Additionally, under hypoxic conditions, HIF-1α-induced H19 expression activates the AMP-activated protein kinase pathway, which promotes lipid droplet degradation in HSCs and encourages their activation (Tian et al., 2021; Gao and Mao, 2024). CircRNAs play an important role in liver fibrosis by regulating HSCs and other liver cells. They can directly stimulate Kupffer cells (KCs) to release chemokines and inflammatory cytokines, which then activate HSCs. CircMcph1, which worsens fibrosis by acting as a sponge for miR-370-3p, is a notable example. This sequestration increases Irak2 expression, enhancing KC-mediated inflammation and leading to the indirect activation of HSCs (Xu et al., 2023). Alcohol, drugs, and parasites can also influence HSC activation. Alcohol affects HSCs by altering the chromatin structure, which increases the expression of ECM proteins. It also promotes elastin accumulation in HSCs by stimulating tropoelastin gene transcription, elastin protein production, and TIMP-1 gene transcription (Page et al., 2015; Higashi et al., 2017). Long-term methotrexate (MTX) therapy poses a significant risk of liver damage. Its metabolite, MTX-polyglutamate, inhibits 5-aminoimidazole-4-carboxamide ribonucleotide transformylase, causing adenosine buildup inside cells. This activates HSCs, enhances ECM accumulation, and causes liver fibrosis (Ezhilarasan, 2021). (Fig. 3)
Fig. 3. Extracellular factors promoting HSC activationhepatic stellate cell (HSC) activation. Chemokine roles in HSC activationThe term "chemokine" originates from "chemotactic cytokines," emphasizing their primary role in guiding cell movement (Charo and Ransohoff, 2006; Marra and Tacke, 2014). These small proteins (6–14 kDa) are rich in basic amino acids and can bind to heparin, playing an essential role in immune cell trafficking (Kim and Broxmeyer, 1999; Rossi and Zlotnik, 2000). Chemokines influence tissue epithelium, growth, and blood vessel formation in addition to their chemotactic function (Inagaki and Okazaki, 2007). Chemokines serve as key mediators in activating HSCs and maintaining their fibrogenic phenotype (Saiman and Friedman, 2012; Seki and Brenner, 2015). The transition of HSCs from a resting to an active state is crucial for fibrogenesis (Mederacke et al., 2013; Kisseleva and Brenner, 2021). Many chemokines either target or are produced by HSCs, thus creating a profibrotic environment. Several members of the CC chemokine family are involved in HSC activation and fibrosis. For example, CCL2, which is released by various cell types in the inflamed liver, promotes HSC migration and activation (Marra 1999; Ramm, 2009). CCL5, which increases in fibrotic livers, aids in activating HSCs and promoting fibrosis. Studies have shown that blocking CCL5 or its receptor CCR5 reduces experimental fibrosis (Seki et al., 2009; Berres et al., 2010; Heinrichs et al., 2013). Similarly, CCL20, which is produced by damaged hepatocytes and cholangiocytes, is highly expressed in alcoholic hepatitis and enhances hepatocyte stem cell (HSC)-driven fibrogenesis (Schwabe et al., 2003; Affò et al., 2014). In the CXC chemokine family, CXCL10 directly promotes fibrogenesis in HSCs while inhibiting natural killer (NK) cell-driven HSC inactivation (Hintermann et al., 2010). Interestingly, HSCs also produce CXCL9 and CXCL10, with CXCL9 potentially exerting antifibrotic effects (Tacke et al., 2011; Sahin et al., 2012). Monocytes and macrophages, which are recruited by chemokines such as CCL1 and CCL2 and members of the growth-regulated oncogene (chemokine CXCL1) family, play dual roles in liver fibrosis. Although they can promote tissue repair, they also worsen inflammation and fibrosis by secreting chemokines that further activate HSCs (Pellicoro et al., 2014). Intracellular signaling pathways in the activation of HSCTransforming growth factor-beta signaling is the key pathway that drives HSC activation and ECM production. In healthy livers, quiescent HSCs (Q-HSCs) produce minimal TGF-β, but their levels increase quickly after liver injury (Carthy, 2018). Activated HSCs generate TGF-β, creating a positive feedback loop that enhances fibrogenesis through SMAD2/SMAD3 signaling, while SMAD7 functions as an inhibitory regulator (Shi et al., 2011). TLR4 activation in Q-HSCs decreases the TGF-β pseudo-receptor BAMBI, boosting HSC responsiveness to TGF-β (Seki et al., 2007). Additionally, macrophages that engulf apoptotic bodies secrete TGF-β, further activating HSCs (Kisseleva and Brenner, 2006; Li et al., 2009). The Notch signaling pathway, which mediates cell-to-cell communication through ligand-receptor interactions, is also involved in the activation of HSCs. In mammals, four Notch receptors (Notch1–4) and five ligands (Jagged1, Jagged2, Delta-like 1, 3, and 4) regulate tissue development (Meurette and Mehlen, 2018). Sawitza et al. (2009) provided initial evidence that JAG1 enhances the expression of α-SMA and collagen in HSCs. This role of the Notch pathway has been supported by subsequent research (Cong et al., 2021). In HSCs activated by TGF-β, fibrosis-related genes (e.g., collagen I and α-SMA) and components of the Notch pathway (e.g., Notch3, JAG1, and Hes1) are upregulated (Bansal et al., 2015). Notch signaling directly activates HSCs and promotes fibrosis through interactions with neighboring cells, such as LSECs (Duan et al., 2018). The Wnt/β-catenin pathway, crucial for embryogenesis and cell organization, also plays a role in liver fibrosis. Genomic analyses of liver samples from patients with primary biliary cirrhosis have shown increased levels of Wnt pathway components (Schunk et al., 2021). Specifically, Wnt5a and its receptor Frizzled-2 are highly expressed during HSC differentiation into myofibroblasts, emphasizing their importance in fibrosis development (Shackel, 2001; Jiang et al., 2006). The Hedgehog (Hh) pathway is a complex and evolutionarily conserved signaling cascade that contributes to HSC activation and their transition into myofibroblasts (Omenetti et al., 2011; Bangs and Anderson, 2017). Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ), mechanosensitive transcriptional regulators, play a critical role in maintaining fibrosis by amplifying matrix-driven feedback loops (Liu et al., 2015). YAP/TAZ levels increase in hepatocytes in both mouse and human liver injury models, attracting macrophages and promoting inflammation and fibrosis (Mooring et al., 2020). YAP inhibition prevents HSC activation and decreases the expression of fibrogenic markers such as α-SMA and type I collagen (Mannaerts et al., 2015) (Fig. 4).
Fig. 4. Intracellular signaling pathways involved in the activation of hepatic stellate cells Multiple intracellular pathways regulate hepatic stellate cell activation, proliferation, and fibrogenic responses. Role of hepatic stellate cells in liver diseasesHSC activation and fibrosis are key to disease progression in non-alcoholic fatty liver disease (NAFLD). Multiple mechanisms drive the shift from simple steatosis to nonalcoholic steatohepatitis (NASH) and fibrosis. Both intrahepatic and extrahepatic factors, including inflammation and lipotoxicity, activate HSCs. Important molecules involved in this process include PPARγ, adiponectin receptor 1, perilipin 2 (PLIN2/ADFP), C/EBPα, C/EBPβ, and SREBP-1c, all of which are expressed by HSCs (Kisseleva et al., 2012). Leptin, an adipocyte-derived hormone, further mediates HSC activation in obesity (Potter et al., 2003). The development of NASH may also involve free fatty acids (FFAs) from triglyceride breakdown and fat tissue, as well as new fat creation. Their excessive buildup triggers lipogenesis-related inflammasome activation in liver cells, stress in the ER, and oxidative stress. These cellular disruptions ultimately cause liver cell death and the release of cytokines and chemokines, which activate HSCs. This, in turn, promotes liver fibrosis involving various immune cells, such as macrophages, neutrophils, lymphocytes, and Kupffer cells (Fig. 5) (Wang et al., 2022).
Fig. 5. Proposed mechanisms of NASH pathogenesis involving FFAs and HSCs. Gut-derived microbial products, such as LPS, reach the liver through the portal vein and stimulate TLRs, especially TLR4, on liver cells (Ganz and Szabo, 2013). Higher serum LPS levels in patients with NASH and animal models highlight the role of TLR4 in fibrosis (Miele et al., 2009). When TLR4 is activated, it downregulates the TGF-β pseudo-receptor BAMBI in HSCs, thereby increasing ECM production (Seki et al., 2007). Activated HSCs also contribute to liver inflammation by secreting cytokines and expressing multiple TLRs (Seki and Schwabe, 2015; Schuster et al., 2018). HSC activation is a key point in NASH development, with transcriptional regulators such as growth-regulated oncogene (chemokine CXCL1) and runt-related transcription factor 1 (RUNX1) playing important roles (Breuer et al., 2020). Additionally, CD8+ T cells interact with HSCs to promote inflammation in NASH livers (Breuer et al., 2020). Motif enrichment analyses at promoter regions of HSC-activated genes indicate that ETS1 and RUNX1 are putative transcriptional regulators driving NASH-associated HSC plasticity. ETS1 is a transcription factor that transmits growth factor and extracellular signal-regulated kinase signaling, regulating genes involved in key processes of HSC activation: inflammation, proliferation, and ECM remodeling. The role of ETS1 in NASH-associated HSC plasticity is consistent with the central importance of TGFβ signaling in HSC activation, as ETS1 is known to be permissive for TGFβ-Smad2/3 signaling in other cell types. A central finding is that ETS1 likely acts cooperatively with RUNX1. Although the broad induction of genes with ETS1 motifs suggests that it functions as a transcriptional amplifier, the presence of both ETS1 and RUNX1 motifs is a stronger predictor of gene induction during HSC activation (Marcher et al., 2019). This cooperation is thought to define a transcriptional program for signature processes such as cell adhesion, motility, and proliferation. The interaction may involve RUNX1 stabilizing higher-order transcription factor complexes at proximal ETS1 binding sites rather than composite DNA binding sites (Mehal, 2023). Furthermore, CD4+ T cell subsets are important drivers of NAFLD progression from steatosis to fibrosis (Her et al., 2020; Mehal, 2023). Fibrosis-specific evidence indicates that CD8+ T cells can activate HSCs in vitro and in vivo via an IL-10 mechanism (Breuer et al., 2020; Mehal, 2023). Patients with NAFLD and advanced fibrosis have a 3-fold higher risk of all-cause mortality and 10-fold higher risk of liver-related mortality than patients without advanced fibrosis, underscoring HSC activation as a key therapeutic target (Taylor et al., 2020; Ng et al., 2023) Liver fibrosis, cirrhosis, and hepatocellular carcinomaLiver fibrosis occurs due to excessive ECM accumulation, which damages tissue structure and impairs liver function (Henderson et al., 2020). Ongoing liver injury leads to chronic activation of HSCs, continuous ECM deposition, and ultimately cirrhosis (Hammerich and Tacke, 2023; Wiering et al., 2023). TGF-β serves as the main driver of this imbalance between ECM creation and degradation (Kim et al., 2022; Wiering et al., 2023). Fibrosis typically develops over years or even decades following persistent liver damage and is unexpectedly associated with a robust immune response (Kisseleva and Brenner, 2021). Liver fibrosis is characterized by the buildup of collagen and other fibrillar proteins, such as elastin, in the space of Disse, along with increased ECM proteins (Higashi et al., 2017). Excess ECM distorts the liver’s architecture, impairs its functions, affects blood flow, and can lead to cirrhosis (Senoo et al., 2010). TGF-β is key in the imbalance between ECM production and degradation that occurs in liver fibrosis (Hammerich and Tacke, 2023; Wiering et al., 2023). Activated HSCs produce αSMA, collagen I, matrix degradation inhibitors, proinflammatory cytokines, and chemokines that draw immune cells to the inflammation site through positive chemotaxis (Hammerich and Tacke, 2023; Wiering et al., 2023). These activated HSCs exhibit a contractile, proliferative, and fibrogenic phenotype (Kisseleva and Brenner, 2021; Lee and Seki, 2023). Ultrastructurally, they are marked by a rough endoplasmic reticulum and a Golgi apparatus responsible for collagen production (Lee and Seki, 2023; Wiering et al., 2023). Additionally, they facilitate immune cell recruitment and infiltration into the liver by releasing proinflammatory cytokines such as CCL2, CCL5, IL8, and CXCL12 and by expressing adhesion molecules like ICAM-1 and VCAM-1 (Kisseleva and Brenner, 2021; Hammerich and Tacke, 2023; Wiering et al., 2023). These cells migrate to injury sites, aiding in the formation of fibrous scars. HSCs, LSECs, and hepatic macrophages constitute the liver microcirculatory environment, which plays a vital role in maintaining vascular tone and managing inflammation (Ganz and Szabo, 2013). Growth factors such as TGF-β, PDGF, and EGF, significantly promote HSC activation (Friedman et al., 2018; Kisseleva and Brenner, 2021; Subramanian et al., 2022; Wiering et al., 2023; Hammerich and Tacke, 2023). Once activated, HSCs can produce autocrine signals that reinforce their fibrogenic activity (Friedman et al., 2018; Schwabe et al., 2020). Myofibroblasts play a vital role in fibrotic diseases affecting the lungs, kidneys, and liver (Friedman et al., 2013). They are the main cells that produce ECM, including collagen I and III, during liver fibrosis. Although the origins of myofibroblasts have been debated, previous studies have indicated that their primary sources are HSCs and portal myofibroblasts. Myofibroblasts develop from activated HSCs in response to various cytokines and inflammatory cells following liver injury. Excess cytokines can keep myofibroblasts activated, resulting in the production of large amounts of ECM. Portal myofibroblasts are the main source of myofibroblasts in biliary disease (Iwaisako et al., 2014; Wells and Schwabe, 2015). Additionally, animal studies suggest that HSCs and myofibroblasts can also originate from mesothelial cells via mesothelial-mesenchymal transition after liver injury (Li et al., 2013). Genetic studies using LratCre-based models have demonstrated an overall pro-tumoral role for HSCs during the development of HCC (Otto et al., 2023). However, HSCs can also exhibit antitumoral properties under specific conditions, reflecting their complex and context-dependent functions (Filliol et al., 2022; Sererols-Viñas et al., 2025). In NAFLD/NASH, HSCs are activated by metabolic stress, leptin, and LPS, leading them to produce collagen and inflammatory mediators such as TGF-β and CCL2. In advanced stages, such as fibrosis and HCC, activated HSCs further drive disease progression by secreting a range of pro-fibrotic factors and signals that remodel the liver environment (Table 1). Table 1. Key functions of hepatic stellate cells in different liver diseases (authors’ synthesis).
Therapeutic options targeting HSC activationHSCs are a key target for anti-fibrotic treatments due to their crucial role in liver fibrosis. Current therapies aim to regulate metabolism, protect hepatocytes, and modulate the immune response. PPAR agonists, farnesoid X receptor agonists, and glucagon-like peptide agonists target metabolic dysfunction, whereas CCR2/5 antagonists and apoptosis signal-regulating kinase 1 (ASK-1) inhibitors address inflammation and immune cell recruitment (Seki et al., 2009). These strategies indirectly decrease HSC activation by targeting upstream mediators. Nuclear receptors, such as PPAR and FXR, expressed in HSCs help suppress their activation, as studies indicate that PPARγ and FXR agonists, such as GW570 (Krützfeldt et al., 2005; Janssen et al., 2013), pioglitazone (Krützfeldt et al., 2005; Janssen et al., 2013), and obeticholic acid (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015), can induce HSC senescence, thereby reducing liver fibrosis severity. TK (tyrosine kinase) is present in HSCs, and its activation leads to their transformation into an active state; therefore, TK inhibition might be a promising approach for treating liver fibrosis. Sorafenib, which is used in patients with HCC, has been shown to decrease cirrhosis complications such as portal hypertension and has demonstrated anti-fibrotic effects in numerous trials (Ma et al., 2017). Nilotinib inhibits histone deacetylase and promotes apoptosis and autophagic cell death in activated HSCs (Krützfeldt et al., 2005; Janssen et al., 2013). However, most of these drugs are still in animal studies and not yet available clinically; their safety and effectiveness require further investigation (Table 2). Table 2. Clinical efficacy of selected investigational therapies for liver fibrosis (adapted from Nasir et al., 2025 ).
Direct HSC-targeting therapies are also being studied. For example, CCR5 inhibitors, such as maraviroc, block CCL5-mediated HSC activation (Lee and Seki, 2023). Galectin-3 inhibitors (e.g., belapectin) and HSP47 siRNA nanoparticles (e.g., BMS-986263) can reduce fibrosis by targeting ECM production and collagen stabilization (Ito and Nagata, 2019; Slack et al., 2021). Additionally, lysyl oxidase inhibitors (e.g., simtuzumab) and integrin-αV blockers reduce fibrosis in preclinical models (Henderson et al., 2013; Ikenaga et al., 2017). Angiotensin receptor antagonists, known for their heart benefits, also inhibit HSC activation in animal studies, though human trials are still needed (Li et al., 2017). Table 2 summarizes the results from recent clinical trials investigating novel therapies for NASH. These include belapectin, obeticholic acid, and a combination of selonsertib, cilofexor, and firsocostat (Table 2) (Nasir et al., 2025). Despite these advances, specific antifibrotic therapies remain limited. Ongoing research, particularly single-cell analyses, may provide new insights into the biology of HSCs and pave the way for innovative treatments. Future perspectivesHepatic stellate cells are key players in liver fibrosis development. They are the primary source of ECM-producing myofibroblasts. Their activation is driven by a complex mix of signals. These include inflammatory, metabolic, and mechanical signals. This activation is a characteristic feature of chronic liver diseases, including NAFLD, alcoholic hepatitis, and viral hepatitis. This review has synthesized the vast knowledge on HSC biology, from fundamental mechanisms to emerging therapeutic strategies, providing a comprehensive resource for researchers and clinicians alike. In recent decades, significant progress has been made. We now understand the molecular mechanisms behind HSC activation, such as the roles of chemokines, intracellular signaling pathways (e.g., TGF-β, Notch, Wnt/β-catenin, and Hedgehog), and transcriptional regulators (e.g., YAP/TAZ). These findings have enhanced our understanding of liver fibrosis (Intercept Pharmaceuticals, Inc., 2022). Despite these advances, several challenges remain. First, the heterogeneity of HSCs is not fully understood. Single-cell RNA sequencing and spatial transcriptomics are promising tools. They explored the diversity of HSC populations and their interactions with other liver cell types, such as hepatocytes, Kupffer cells, and LSECs. These technologies could uncover new HSC subpopulations with distinct roles in fibrosis progression or regression, paving the way for more precise therapeutic strategies. Second, although many antifibrotic agents have shown promise in preclinical models, their translation into clinical practice remains limited. Many therapies targeting HSCs or their signaling pathways face hurdles related to specificity, effectiveness, and safety. Broad inhibition of TGF-β signaling, while effective in reducing fibrosis, may interfere with its vital roles in tissue homeostasis and immune regulation. Similarly, targeting chemokine receptors such as CCR5 or integrins like αVβ6 requires careful evaluation. Their functions in other organs should be considered. The development of cell-specific delivery systems, such as nanoparticles or ligand-conjugated drugs, could improve the precision of antifibrotic therapies and reduce off-target effects. The bidirectional relationship between HSCs and the immune system presents both challenges and opportunities. Macrophages and T cells stimulate HSC activation through secretion of cytokines and chemokines. Activated HSCs can influence immune responses by expressing antigen-presenting molecules or releasing immunoregulatory factors. Leveraging this interaction to promote fibrosis resolution while preventing excessive inflammation or immunosuppression is a key focus of future research. Immunotherapies, such as checkpoint inhibitors or adoptive cell transfer, could be combined with antifibrotic agents to produce synergistic effects. Finally, developing reliable biomarkers for HSC activation and fibrosis progression is essential for early diagnosis, patient stratification, and treatment outcome monitoring. Current noninvasive methods, such as elastography and serum biomarkers (e.g., ELF test, PRO-C3), have limitations in sensitivity and specificity. Combining multi-omics techniques (e.g., proteomics, metabolomics) with advanced imaging could lead to the identification of new biomarkers that mirror the dynamic state of HSCs and the fibrotic microenvironment. Recent research has identified candidate serum biomarkers at various stages of liver disease, including inflammatory cytokines, fatty acid transporter proteins, lipid droplet-associated proteins, and ECM proteins. Recent improvements, including the NIS2 + ™ panel, have shown the potential to further reduce unnecessary biopsies and screening costs in clinical practice and trials. The integration of multi-omics data (proteomics, metabolomics) with advanced imaging is essential for creating dynamic biomarker panels. Future clinical trials must adopt a precision medicine approach that stratifies patients according to their underlying fibrosis phenotype. ConclusionIn summary, while progress has been made in understanding HSCs and liver fibrosis, more work is needed to develop effective treatments. Future research should focus on HSC heterogeneity, targeted antifibrotic therapies, and the use of the immune system for fibrosis resolution. An alternative approach is RNA interference (RNAi), a promising gene therapy technology inspired by microRNAs that reduces gene expression by targeting mRNA with small interfering RNAs. RNAi has applications in research, therapy, and treatment of neurodegenerative diseases, genetic disorders, viral infections, and cancers. Like some drugs, such as Inclisiran, which is an RNAi-based drug approved for the treatment of hypercholesterolemia by targeting the mRNA of the PCSK9 gene, preventing the production of the PCSK9 protein. It is used to lower the LDL cholesterol level. By addressing these challenges, the ultimate goal of preventing, halting, or even reversing liver fibrosis can be achieved, thereby improving outcomes for patients with chronic liver diseases. Limitations and prospectsWhile this review summarizes the current HSC biology, its limitations offer research opportunities. A key challenge is reliance on rodent models and in vitro cultures, which do not fully reflect the complexity and chronicity of human liver disease. Most mechanistic studies view HSCs as uniform, but single-cell data now show significant heterogeneity. To fill these gaps and turn knowledge into treatments, future research should focus on the following areas: -Decoding HSC heterogeneity requires more research, especially on human subpopulations. Spatial transcriptomics and multiplexed imaging are vital for mapping these subsets in the liver and understanding their roles in the development and regression of fibrosis. -Bridging the translational gap requires human-relevant models, as emphasized by the high antifibrotic trial failure rate. The development of targeted delivery systems, such as nanoparticles with HSC-specific ligands, is key to improving drug efficacy and safety by minimizing off-target effects like TGF-β. -Leveraging the immune-stromal axis: The crosstalk between HSCs and immune cells offers a promising but complex therapeutic frontier. Future research should focus on manipulating this interaction, such as targeting specific HSC subpopulations that modulate immune responses, to resolve fibrosis without causing excessive immunosuppression or inflammation. -Developing dynamic biomarkers: There is a critical need for biomarkers that reflect HSC activation states and treatment response beyond static fibrosis measurement. Integrating multi-omics approaches (proteomics, metabolomics) with clinical data to identify HSC-specific serum markers will enable better patient stratification and monitoring in clinical trials. Focusing on these areas allows future research to overcome current limitations and develop effective, targeted antifibrotic therapies. List of abbreviationsα-SMA, Alpha-Smooth Muscle Actin; aHSCs, Activated hepatic stellate cells; CL, Chemokine (C-C motif) ligand; CXCL, Chemokine (C-X-C motif) Ligand; DAMP, Damage-Associated Molecular Patterns; ECM, Extracellular matrix; FFAs, Free fatty acids; FXR, Farnesoid X receptor; GFAP, Glial fibrillary acidic protein; GLUT, Glucose transporter; HCC, Hepatocellular carcinoma; Hh, Hedgehog; HSCs, Hepatic Stellate Cells; IFN-γ, Interferon Gamma; IL, Interleukin; LPS, Lipopolysaccharide; LRAT, Lecithin:retinol acyltransferase; LSECs, Liver Sinusoidal Endothelial Cells; LXRα, Liver X receptor alpha; MCP-1, Monocyte Chemoattractant Protein-1; M-CSF, Macrophage Colony-Stimulating Factor; MFs, Myofibroblasts; MIP-2, Macrophage Inflammatory Protein-2; miRNA, microRNA; MMPs, Matrix metalloproteinases; NAFLD, Non-Alcoholic Fatty Liver Disease; NASH, Non-Alcoholic Steatohepatitis; NFκB, Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells; NGF, Nerve growth factor; NK cells, natural killer cells; NO, Nitric oxide; PAMP, Pathogen-Associated Molecular Patterns; PDGF, Platelet-Derived Growth Factor; PKM2, Pyruvate Kinase M2; PPAR, Peroxisome Proliferator-Activated Receptor; qHSCs, Quiescent hepatic stellate cells; RAR, Retinoic acid receptor; RBP, Retinol-Binding Protein; ROS, Reactive oxygen species; rER, Rough endoplasmic reticulum; RXR, Retinoid X receptor; scRNA-seq, Single-Cell RNA Sequencing; SREBP-1c, Sterol Regulatory Element-Binding Protein-1c; TGF-β, Transforming Growth Factor-Beta; TIMP, Tissue inhibitor of metalloproteinases; TLRs, Toll-Like Receptors; TNF, Tumor necrosis factor; YAP/TAZ, Yes-Associated Protein/Transcriptional Co-activator with PDZ-Binding Motif. Conflicts of interestThe authors have no conflicts of interest to declare. FundingThis study received no specific funding. Author’s contributionsHE, MN, MM: Acquisition, analysis, and interpretation of the data, writing of the original draft, review, and editing. YN, RE, NZ, OE, and MAL: Revision and interpretation of data, review, and editing. HE, YN, RE, NZ, OE, and MAL: revision and interpretation of data, review, and editing. HE, MN, and MM: supervised the work, contributed to manuscript writing, analysis and interpretation of data, preparation of figures, review, and editing, and final approval of the manuscript. ReferencesAffò, S., Morales-Ibanez, O., Rodrigo-Torres, D., Altamirano, J., Blaya, D., Dapito, D.H., Millán, C., Coll, M., Caviglia, J.M., Arroyo, V., Caballería, J., Schwabe, R.F., Ginès, P., Bataller, R. and Sancho-Bru, P. 2014. CCL20 mediates lipopolysaccharide induced liver injury and is a potential driver of inflammation and fibrosis in alcoholic hepatitis. Gut 63, 1782–1792; doi:10.1136/gutjnl-2013-306098 Ajat, M., Molenaar, M., Brouwers, J.F.H.M., Vaandrager, A.B., Houweling, M. and Helms, J.B. 2017. Hepatic stellate cells retain the capacity to synthesize retinyl esters and to store neutral lipids in small lipid droplets in the absence of LRAT. Biochim. Biophys. Acta. Mol. Cell. Biol. Lipids 1862, 176–187. doi: 10.1016/j.bbalip.2016.10.013 Alexandra, B., Asha, B., Michael, O. and Amar D.S. 2024. “The Good, the Bad, and the Ugly” – about diverse phenotypes of hepatic stellate cells in the liver. Cell. Mol. Gastroenterol. Hepatol. 17(4), 607–622, doi: 10.1016/j.jcmgh.2024.01.002. Asahina, K., Zhou, B., Pu, W.T. and Tsukamoto, H. 2011. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 53(3), 983–995; doi:10.1002/hep.24149 Baba, S., Fujii, H., Hirose, T., Yasuchika, K., Azuma, H., Hoppo, T., Naito, M., Machimoto, T. and Ikai, I. 2004. Commitment of bone marrow cells to hepatic stellate cells in mouse. J. Hepatol. 40, 255–260; doi:10.1016/j.jhep.2003.10.012 Baggiolini, M. 1998. Chemokines and leukocyte traffic. Nature 392, 565–568; doi:10.1038/33340 Bangs, F. and Anderson, K.V. 2017. Primary cilia and mammalian hedgehog signaling. Cold. Spring. Harb. Perspect. Biol. 9(9), 28175; doi:10.1101/cshperspect.a028175 Bansal, R., Van Baarlen, J., Storm, G. and Prakash, J. 2015. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 5, 18272; doi:10.1038/srep18272 Bartel, D.P. and Chen, C.Z. 2004. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 5, 396–400; doi:10.1038/nrg1328 Bataller, R. and Brenner, D.A. 2005. Liver fibrosis. J. Clin. Invest. 115(2), 209–218; doi:10.1172/JCI24282 Berres, M.L., Koenen, R.R., Rueland, A., Zaldivar, M.M., Heinrichs, D., Sahin, H., Schmitz, P., Streetz, K.L., Berg, T., Gassler, N., Weiskirchen, R., Proudfoot, A., Weber, C., Trautwein, C. and Wasmuth, H.E. 2010. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J. Clin. Invest. 120, 4129–4140; doi: 10.1172/JCI41732 Blomhoff, R., Green, M.H., Berg, T. and Norum, K.R. 1990. Transport and storage of vitamin A. Science 250, 399–404; doi:10.1126/science.2218545 Blomhoff. 1994. Vitamin A in health and disease. New York: Marcel Dekker; doi:10.1201/9781482277562 Breuer, D.A., Pacheco, M.C., Washington, M.K., Montgomery, S.A., Hasty, A.H. and Kennedy, A.J. 2020. CD8+ T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver. Physiol. 318(2), G211–G224; doi: 10.1152/ajpgi.00040.2019 Carthy, J.M. 2018. TGFβ signaling and the control of myofibroblast differentiation: implications for chronic inflammatory disorders. J. Cell. Physiol. 233, 98–106; doi:10.1002/jcp.25879 Casini, A. 1997. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: role of nitric oxide. Hepatology 25, 361–367; doi:10.1002/hep.510250218 Cassiman, D., Barlow, A., Borght, S.V., Libbrecht, L. and Pachnis, V. 2006. Hepatic stellate cells do not derive from the neural crest. J. Hepatol. 44, 1098–1104; doi:10.1016/j.jhep.2005.09.023 Cassiman, D., Libbrecht, L., Desmet, V., Denef, C. and Roskams, T. 2002. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J. Hepatol. 36(2), 200–209; doi:10.1016/S0168-8278(01)00260-4 Cassiman, D., Van Pelt, J., De Vos, R., Van Lommel, F., Desmet, V., Yap, S.H. and Roskams, T. 1999. Synaptophysin: a novel marker for human and rat hepatic stellate cells. Am. J. Pathol. 155, 1831–1839; doi:10.1016/S0002-9440(10)65501-0 Chan, K.M., Fu, Y.H., Wu, T.J., Hsu, P.Y. and Lee, W.C. 2013. Hepatic stellate cells promote the differentiation of embryonic stem cell derived definitive endodermal cells into hepatic progenitor cells. Hepatol. Res. 43, 648–657; doi:10.1111/hepr.12000 Charo, I.F. and Ransohoff, R.M. 2006. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621; doi:10.1056/NEJMra052723 Chawla, A., Repa, J.J., Evans, R.M. and Mangelsdorf, D.J. 2001. Nuclear receptors and lipid physiology: opening the X-files. Science 294(5548), 1866–1870; doi:10.1126/science Chen, Y., Choi, S.S., Michelotti, G.A., Chan, I.S., Swiderska-Syn, M., Karaca, G.F., Xie, G., Moylan, C.A., Garibaldi, F., Premont, R., Suliman, H.B., Piantadosi, C.A. and Diehl, A.M. 2012. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 143, 1319–1329; doi:10.1053/j.gastro.2012.07.115 Coll, M., Taghdouini, A.E., Perea, L., Mannaerts, I., Vila-Casadesús, M., Blaya, D., Rodrigo-Torres, D., Affò, S., Morales-Ibanez, O., Graupera, I., Lozano, J.J., Najimi, M., Sokal, E., Lambrecht, J., Ginès, P., Van Grunsven, L.A. and Sancho-Bru, P. 2015. Integrative miRNA and gene expression profiling analysis of human quiescent hepatic stellate cells. Sci. Rep. 5, 11549; doi:10.1038/srep11549 Cong, S., Liu, Y., Li, Y., Chen, Y., Chen, R., Zhang, B., Yu, L., Hu, Y., Zhao, X., Mu, M., Cheng, M. and Huang, Z. 2021. MiR-571 affects the development and progression of liver fibrosis by regulating the Notch3 pathway. Sci. Rep. 11, 21854; doi: 10.1038/s41598-021-00638-3 Coulon, S., Heindryckx, F., Geerts, A., Van Steenkiste, C., Colle, I. and Van Vlierberghe, H. 2011. Angiogenesis in chronic liver disease and its complications. Liver. Int. 31(2), 146–162; doi:10.1111/j.1478-3231.2010.02369.x Coulouarn, C. and Clément, B. 2014. Stellate cells and the development of liver cancer: therapeutic potential of targeting the stroma. J. Hepatol. 60(6), 1306–1309; doi:10.1016/j.jhep.2014.02.013 De Minicis, S., Rychlicki, C., Agostinelli, L., Saccomanno, S., Candelaresi, C., Trozzi, L., Mingarelli, E., Facinelli, B., Magi, G., Palmieri, C., Marzioni, M., Benedetti, A. and Svegliati-Baroni, G. 2014. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 59, 1738–1749; doi:10.1002/hep.26695 Denby, L., Ramdas, V., Lu, R., Conway, B.R., Grant, J.S., Dickinson, B., Aurora, A.B., McClure, J.D., Kipgen, D., Delles, C., Van Rooij, E. and Baker, A.H. 2014. MicroRNA-214 antagonism protects against renal fibrosis. J. Am. Soc. Nephrol. 25(1), 65–80; doi:10.1681/ASN.2013010072 Dieter, P., Scheibe, R., Jakobsson, P.J., Watanabe, K., Kolada, A. and Kamionka, S. 2000. Functional coupling of cyclooxygenase 1 and 2 to discrete prostanoid synthases in liver macrophages. Biochem. Biophys. Res. Commun. 276, 488–492; doi:10.1006/bbrc.2000.3496 Dooley, S., Delvoux, B., Lahme, B., Mangasser-Stephan, K. and Gressner, A.M. 2000. Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology 31, 1094–1106; doi:10.1053/he.2000.6126 Du, D., Liu, C., Qin, M., Zhang, X., Xi, T., Yuan, S., Hao, H. and Xiong, J. 2022. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta. Pharm. Sin. B. 12, 558–580; doi:10.1016/j.apsb.2021.09.019 Du, K., Jun, J.H., Dutta, R.K. and Diehl, A.M. 2024. Plasticity, heterogeneity, and multifunctionality of hepatic stellate cells in liver pathophysiology. Hepatol. Commun. 8(5), 41; doi:10.1097/HC9.0000000000000411 Duan, J.L., Ruan, B., Yan, X.C., Liang, L., Song, P., Yang, Z.Y., Liu, Y., Dou, K.F., Han, H. and Wang, L. 2018. Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 68, 677–690; doi:10.1002/hep.29834 Enzan, H., Hayashi, Y., Miyazaki, E., Naruse, K., Tao, R., Kuroda, N., Nakayama, H., Kiyoku, H., Hiroi, M. and Saibara, T. 1999. Morphological aspects of hepatic fibrosis and Ito cells (hepatic stellate cells), with special reference to their myofibroblastic transformation.In Liver Diseases and Hepatic Sinusoidal Cells. Tanikawa. and Ueno Tokyo, Japan: Springer, pp: 219–31. doi: 10.1007/978-4-431-67935-6_18 Ezhilarasan, D. 2020. MicroRNA interplay between hepatic stellate cell quiescence and activation. Eur. J. Pharmacol. 885, 173507; doi:10.1016/j.ejphar.2020.173507 Ezhilarasan, D. 2021. Hepatotoxic potentials of methotrexate: understanding the possible toxicological molecular mechanisms. Toxicology 458, 152840. doi: 10.1016/j.tox.2021.152840 Filliol, A., Saito, Y., Nair, A., Dapito, D.H., Yu, L.X., Ravichandra, A., Bhattacharjee, S., Affo, S., Fujiwara, N., Su, H., Sun, Q., Savage, T.M., Wilson-Kanamori, J.R., Caviglia, J.M., Chin, L., Chen, D., Wang, X., Caruso, S., Kang, J.K., Amin, A.D., Wallace, S., Dobie, R., Yin, D., Rodriguez-Fiallos, O.M., Yin, C., Mehal, A., Izar, B., Friedman, R.A., Wells, R.G., Pajvani, U.B., Hoshida, Y., Remotti, H.E., Arpaia, N., Zucman-Rossi, J., Karin, M., Henderson, N.C., Tabas, I. and Schwabe, R.F. 2022. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature 610, 356–365; doi: 10.1038/s41586-022-05289-6 Friedman, S.L. 2000. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 275, 2247–2250; doi:10.1074/jbc.275.4.2247 Friedman, S.L. and Arthur, M.J. 1989. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J. Clin. Invest. 84, 1780–1785; doi:10.1172/JCI114362 Friedman, S.L., Neuschwander-Tetri, B.A., Rinella, M. and Sanyal, A.J. 2018. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–924; doi:10.1038/s41591-018-0104-9 Friedman, S.L., Sheppard, D., Duffield, J.S. and Violette, S. 2013. Therapy for Fibrotic Diseases: nearing the Starting Line. Sci. Transl. Med. 5(167), 167sr1; doi:10.1126/scitranslmed.3004700 Ganz, M. and Szabo, G. 2013. Immune and inflammatory pathways in NASH. Hepatol. Int. 7(S2), 771–781; doi:10.1007/s12072-013-9468-6 Gao, R. and Mao, J. 2024. Noncoding RNA-mediated epigenetic regulation in hepatic stellate cells of liver fibrosis. Noncoding. RNA. 10(4), 44; doi:10.3390/ncrna10040044 Geerts, A. 2001. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver. Dis. 21, 311–335; doi:10.1055/s-2001-17551 Geisler, S., Lichtinghagen, R., Böker, K.H.W. and Veh, R.W. 1997. Differential distribution of five members of the matrix metalloproteinase family and one inhibitor (TIMP-1) in human liver and skin. Cell Tissue Res. 289, 173–183; doi:10.1007/s004410050863 Geng, Y. and Schwabe, R.F. 2025. Hepatic stellate cell heterogeneity: functional aspects and therapeutic implications. Hepatology doi:10.1097/HEP.0000000000001386 Gressner, A.M. 1995. Cytokines an d cellular crosstalk involved in the activation of fat-storing cells. J. Hepatol. 22(Suppl. 2), 28–36. Gressner, A.M. and Haarmann, R. 1988. Regulation of hyaluronate synthesis in rat liver fat storing cell cultures by Kupffer cells. J. Hepatol. 7, 310–318; doi:10.1016/s0168-8278(88)80003-5 Gressner, A.M. and Zerbe, O. 1987. Kupffer cell-mediated induction of synthesis and secretion of proteoglycans by rat liver fat storing cells in culture. J. Hepatol. 5, 299–310; doi:10.1016/S0168-8278(87)80036-3 Gressner, A.M., Lotfi, S., Gressner, G., Haltner, E. and Kropf, J. 1993. Synergism between hepatocytes and Kupffer cells in the activation of fat storing cells (perisinusoidal lipocytes). J. Hepatol. 19, 117–132; doi:10.1016/S0168-8278(05)80185-0 Hagedorn, L., Suter, U. and Sommer, L. 1999. P0 and PMP22 mark a multipotent neural crest-derived cell type that displays community effects in response to TGF-beta family factors. Development 126(17), 3781–3794. Hammerich, L. and Tacke, F. 2023. Hepatic inflammatory responses in liver fibrosis. Nat. Rev. Gastroenterol. Hepatol. 20, 633–646; doi:10.1038/s41575-023-00807-x Heinrichs, D., Berres, M.L., Nellen, A., Fischer, P., Scholten, D., Trautwein, C., Wasmuth, H.E. and Sahin, H. 2013. The chemokine CCL3 promotes experimental liver fibrosis in mice. PLos One. 8(8), e66106; doi:10.1371/journal.pone.0066106 Hellerbrand, C., Jobin, C., Licato, L.L., Sartor, R.B. and Brenner, D.A. 1998. Cytokines induce NF-kappaB in activated but not in quiescent rat hepatic stellate cells. Am. J. Physiol. 275, G269–G278; doi:10.1152/ajpgi.1998.275.2.G269 Henderson, N.C., Arnold, T.D., Katamura, Y., Giacomini, M.M., Rodriguez, J.D., McCarty, J.H., Pellicoro, A., Raschperger, E., Betsholtz, C., Ruminski, P.G., Griggs, D.W., Prinsen, M.J., Maher, J.J., Iredale, J.P., Lacy-Hulbert, A., Adams, R.H. and Sheppard, D. 2013. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 19, 1617–1624; doi:10.1038/nm.3282 Henderson, N.C., Rieder, F. and Wynn, T.A. 2020. Fibrosis: from mechanisms to medicines. Nature 587, 555–566; doi:10.1038/s41586-020-2938-9 Her, Z., Tan, J.H.L., Lim, Y.S., Tan, S.Y., Chan, X.Y., Tan, W.W.S., Liu, M., Yong, K.S.M., Lai, F., Ceccarello, E., Zheng, Z., Fan, Y., Chang, K.T.E., Sun, L., Chang, S.C., Chin, C.L., Lee, G.H., Dan, Y.Y., Chan, Y.S., Lim, S.G., Chan, J.K.Y., Chandy, K.G. and Chen, Q. 2020. CD4(+) T cells mediate the development of liver fibrosis in high fat diet-induced NAFLD in humanized mice. Front. Immunol. 11, 580968; doi:10.3389/fimmu.2020.580968 Herbst, H., Wege, T., Milani, S., Pellegrini, G., Orzechowski, H.D., Bechstein, W.O., Neuhaus, P., Gressner, A.M. and Schuppan, D. 1997. Tissue inhibitor of metalloproteinase-1 and -2 RNA expression in rat and human liver fibrosis. Am. J. Pathol. 150, 1647–1659. Hernández, I., De La Torre, P., Rey-Campos, J., Garcia, I., Sánchez, J.A., Muñoz, R., Rippe, R.A., Muñoz-Yagüe, T. and Solís-Herruzo, J.A. 2000. Collagen alpha1(I) gene contains an element responsive to tumor necrosis factor-alpha located in the 5’ untranslated region of its first exon. DNA Cell Biol. 19, 341–352; doi:10.1089/10445490050043317 Hernandez-Gea, V. and Friedman, S.L. 2011. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 6, 425–456; doi:10.1146/annurev-pathol-011110-130246 Hernández–Gea, V., Ghiassi–Nejad, Z., Rozenfeld, R., Gordon, R., Fiel, M.I., Yue, Z., Czaja, M.J. and Friedman, S.L. 2012. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142, 938–946; doi:10.1053/j.gastro.2011.12.044 Hess, D.C., Abe, T., Hill, W.D., Studdard, A.M., Carothers, J., Masuya, M., Fleming, P.A., Drake, C.J. and Ogawa, M. 2004. Hematopoietic origin of microglial and perivascular cells in brain. Exp. Neurol. 186, 134–144; doi:10.1016/j.expneurol.2003.11.005 Higashi, T., Friedman, S.L. and Hoshida, Y. 2017. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug. Deliv. Rev. 121, 27–42; doi:10.1016/j.addr.2017.05.007 Higashiyama, R., Moro, T., Nakao, S., Mikami, K., Fukumitsu, H., Ueda, Y., Ikeda, K., Adachi, E., Bou–Gharios, G., Okazaki, I. and Inagaki, Y. 2009. Negligible contribution of bone marrow-derived cells to collagen production during hepatic fibrogenesis in mice. Gastroenterology 137, 1459–1466; doi:10.1053/j.gastro.2009.07.006 Hintermann, E., Bayer, M., Pfeilschifter, J.M., Luster, A.D. and Christen, U. 2010. CXCL10 promotes liver fibrosis by prevention of NK cell mediated hepatic stellate cell inactivation. J. Autoimmun. 35, 424–435; doi:10.1016/j.jaut.2010.09.003 Huang, J., Yu, X., Fries, J., Zhang, L. and Odenthal, M. 2014. Microrna function in the profibrogenic interplay upon chronic liver disease. Int. J. Mol. Sci. 15, 9360–9371; doi:10.1002/hep.26695 Ibrahim, S.H., Hirsova, P. and Gores, G.J. 2018. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 67, 963–972; doi:10.1136/gutjnl-2017-315691 Ikenaga, N., Peng, Z.W., Vaid, K.A., Liu, S.B., Yoshida, S., Sverdlov, D.Y., Mikels-Vigdal, A., Smith, V., Schuppan, D. and Popov, Y.V. 2017. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 66, 1697–1708; doi:10.1136/gutjnl-2016-312473 Inagaki, Y. and Okazaki, I. 2007. Emerging insights into Transforming growth factor β Smad signal in hepatic fibrogenesis. Gut 56(2), 284–292; doi:10.1136/gut.2005.088690 Intercept Pharmaceuticals, Inc. 2022. Intercept resubmits new drug application to U.S. FDA for obeticholic acid in patients with liver fibrosis due to NASH. Morristown, NJ: Intercept Pharmaceuticals, Inc. Ito, S. and Nagata, K. 2019. Roles of the endoplasmic reticulum resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease. J. Biol. Chem. 294, 2133–2141; doi:10.1074/jbc.TM118.002812 Iwaisako, K., Jiang, C., Zhang, M., Cong, M., Moore-Morris, T.J., Park, T.J., Liu, X., Xu, J., Wang, P., Paik, Y.H., Meng, F., Asagiri, M., Murray, L.A., Hofmann, A.F., Iida, T., Glass, C.K., Brenner, D.A. and Kisseleva, T. 2014. Origin of Myofibroblasts in the Fibrotic Liver in Mice. Proc. Natl. Acad. Sci. U. S. A. 111(32), E3297–E3305; doi:10.1073/pnas.1400062111 Janssen, H.L.A., Reesink, H.W., Lawitz, E.J., Zeuzem, S., Rodriguez-Torres, M., Patel, K., Van Der Meer, A.J., Patick, A.K., Chen, A., Zhou, Y., Persson, R., King, B.D., Kauppinen, S., Levin, A.A. and Hodges, M.R. 2013. Treatment of HCV Infection by Targeting microRNA. N. Engl. J. Med. 368(18), 1685–1694; doi:10.1056/NEJMoa1209026 Jiang, F., Parsons, C.J. and Stefanovic, B. 2006. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J. Hepatol. 45, 401–409; doi:10.1016/j.jhep.2006.03.016 Kamada, Y., Tamura, S., Kiso, S., Matsumoto, H., Saji, Y., Yoshida, Y., Fukui, K., Maeda, N., Nishizawa, H., Nagaretani, H., Okamoto, Y., Kihara, S., Miyagawa, J.I., Shinomura, Y., Funahashi, T. and Matsuzawa, Y. 2003. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 125, 1796–1807; doi:10.1053/j.gastro.2003.08.029 Karthikeyan, S., Potter, J.J., Geschwind, J.F., Sur, S., Hamilton, J.P., Vogelstein, B., Kinzler, K.W., Mezey, E. and Ganapathy-Kanniappan, S. 2016. Deregulation of energy metabolism promotes antifibrotic effects in human hepatic stellate cells and prevents liver fibrosis in a mouse model. Biochem. Biophys. Res. Commun. 469, 463–469; doi:10.1016/j.bbrc.2015.10.101 Kawada, N., Klein, H. and Decker, K. 1992. Eicosanoid-mediated contractility of hepatic stellate cells. Biochem. J. 285, 367–371; doi:10.1042/bj2850367 Kawada, N., Mizoguchi, Y., Kobayashi, K., Monna, T., Liu, P. and Morisawa, S. 1992. Enhancement of prostaglandin E 2 production by liver macrophages (Kupffer cells) after stimulation with biological response modifiers. Prostaglandins. Leukot. Essent. Fatty Acids 46, 105–110; doi:10.1016/0952-3278(92)90216-6 Kendall, T.J., Duff, C.M., Boulter, L., Wilson, D.H., Freyer, E., Aitken, S., Forbes, S.J., Iredale, J.P. and Hastie, N.D. 2019. Embryonic mesothelial-derived hepatic lineage of quiescent and heterogenous scar-orchestrating cells defined but suppressed by WT1. Nat. Commun. 15(1), 4688; doi:10.1038/s41467-019-12701-9 Kiassov, A.P., Van Eyken, P., Van Pelt, J.F., Depla, E., Fevery, J., Desmet, V.J. and Yap, P.S.H. 1995. Desmin expressing nonhematopoietic liver cells during rat liver development: an immunohistochemical and morphometric study. Differentiation 59, 253–258; doi:10.1046/j.1432-0436.1995.5940253.x Kim, C.H. and Broxmeyer, H.E. 1999. Chemokines: signal lamps for trafficking of T and B cells for development and effector function. J. Leukoc. Biol. 65, 6–15. Kim, E.R., Park, J.S., Kim, J.H., Oh, J.Y., Oh, I.J., Choi, D.H., Lee, Y.S., Park, I.S., Kim, S., Lee, D.H., Cheon, J.H., Bae, J.W., Lee, M., Cho, J.W., An, I.B., Nam, E.J., Yang, S.I., Lee, M.S., Bae, S.H. and Lee, Y.H. 2022. A GLP-1/GLP-2 receptor dual agonist to treat NASH: targeting the gut-liver axis and microbiome. Hepatology 75, 1523–1538; doi:10.1002/hep.32235 Kisseleva, T. and Brenner, D. 2021. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 18, 151–166; doi:10.1038/s41575-020-00372-7 Kisseleva, T. and Brenner, D.A. 2006. Hepatic stellate cells and the reversal of fibrosis. J. Gastroenterol. Hepatol. 21, doi: 10.1111/j.1440-1746.2006.04584.x Kisseleva, T., Cong, M., Paik, Y., Scholten, D., Jiang, C., Benner, C., Iwaisako, K., Moore-Morris, T., Scott, B., Tsukamoto, H., Evans, S.M., Dillmann, W., Glass, C.K. and Brenner, D.A. 2012. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. U. S. A. 109(24), 9448–9453; doi:10.1073/pnas.1201840109 Knittel, T., Müller, L., Saile, B. and Ramadori, G. 1997. Effect of tumour necrosis factor-alpha on proliferation, activation and protein synthesis of rat hepatic stellate cells. J. Hepatol. 27, 1067–1080; doi:10.1016/S0168-8278(97)80151-1 Krützfeldt, J., Rajewsky, N., Braich, R., Rajeev, K.G., Tuschl, T., Manoharan, M. and Stoffel, M. 2005. Silencing of microRNAs In Vivo with ‘antagomirs’. Nature 438(7068), 685–689; doi:10.1038/nature04303 Kupffer, K.V.W. 1876. Ueber Sternzellen der Leber. Briefliche Mitteilung an Prof. Waldyer. Archiv Für Mikroskopische Anatomie 12, 353–358; doi: 10.1007/BF02933897 Lambrecht, J., Mannaerts, I. and Van Grunsven, L.A. 2015. The role of miRNAs in stress-responsive hepatic stellate cells during liver fibrosis. Front. Physiol. 6, 209; doi:10.3389/fphys.2015.00209 Lee, H.J. 2013. Exceptional stories of microRNAs. Exp. Biol. Med. (Maywood). 238(4), 339–343; doi:10.1258/ebm.2012.012251 Lee, U.E. and Friedman, S.L. 2011. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 25(2), 195–206; doi:10.1016/j.bpg.2011.02.005 Lee, Y.S. and Seki, E. 2023. in vivo and in vitro models to study liver fibrosis: mechanisms and limitations. Cell. Mol. Gastroenterol. Hepatol. 16, 355–367; doi:10.1016/j.jcmgh.2023.05.010 Levy, M.T., McCaughan, G.W., Abbott, C.A., Park, J.E., Cunningham, A.M., Müller, E., Rettig, W.J. and Gorrell, M.D. 1999. Fibroblast activation protein: a cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodeling interface in human cirrhosis. Hepatology 29, 1768–1778; doi:10.1002/hep.510290631 Li, A., Zhang, J., Zhang, X., Wang, J., Wang, S., Xiao, X., Wang, R., Li, P. and Wang, Y. 2017. Angiotensin II induces connective tissue growth factor expression in human hepatic stellate cells by a transforming growth factor beta-independent mechanism. Sci. Rep. 7, 7841; doi:10.1038/s41598-017-08334-x Li, C., Kong, Y., Wang, H., Wang, S., Yu, H., Liu, X., Yang, L., Jiang, X., Li, L. and Li, L. 2009. Homing of bone marrow mesenchymal stem cells mediated by sphingosine 1-phosphate contributes to liver fibrosis. J. Hepatol. 50, 1174–1183; doi:10.1016/j.jhep.2009.01.028 Li, Y., Wang, J. and Asahina, K. 2013. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proc. Natl. Acad. Sci. U. S. A. 110(6), 2324–2329. Liu, F., Lagares, D., Choi, K.M., Stopfer, L., Marinković, A., Vrbanac, V., Probst, C.K., Hiemer, S.E., Sisson, T.H., Horowitz, J.C., Rosas, I.O., Fredenburgh, L.E., Feghali-Bostwick, C., Varelas, X., Tager, A.M. and Tschumperlin, D.J. 2015. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung. Cell. Mol. Physiol. 308, L344–L357; doi:10.1152/ajplung.00300.2014 Liu, X., Xu, J., Rosenthal, S., Zhang, L.J., Mccubbin, R., Meshgin, N., Shang, L., Koyama, Y., Ma, H.Y., Sharma, S., Heinz, S., Glass, C.K., Benner, C., Brenner, D.A. and Kisseleva, T. 2020. Identification of lineage-specific transcription factors that prevent activation of hepatic stellate cells and promote fibrosis resolution. Gastroenterology 158, 1728.e1714–1744.e1714; doi:10.1053/j.gastro.2020.01.027 Lua, I. and Asahina, K. 2016. The role of mesothelial cells in liver development, injury, and regeneration. Gut. Liver. 10(2), 166–176; doi:10.5009/gnl15226 Ma, L., Yang, X., Wei, R., Ye, T., Zhou, J.K., Wen, M., Men, R., Li, P., Dong, B., Liu, L., Fu, X., Xu, H., Aqeilan, R.I., Wei, Y.Q., Yang, L. and Peng, Y. 2018. MicroRNA-214 promotes hepatic stellate cell activation and liver fibrosis by suppressing Sufu expression. Cell Death Dis. 9, 718; doi:10.1038/s41419-018-0754-z Ma, R., Chen, J., Liang, Y., Lin, S., Zhu, L., Liang, X. and Cai, X. 2017. Sorafenib: a Potential Therapeutic Drug for Hepatic Fibrosis and its Outcomes. Biomed. Pharmacother. 88, 459–468; doi:10.1016/j.biopha.2017.01.107 Maher, J. 1999. Cytokines: overview. Semin. Liver Dis. 19, 109–115. doi: 10.1055/s-2007-1007103 Mann, J., Chu, D.C.K., Maxwell, A., Oakley, F., Zhu, N.L., Tsukamoto, H. and Mann, D.A. 2010. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 138(2), 705–714; doi:10.1053/j.gastro.2009.10.002 Mann, J., Oakley, F., Akiboye, F., Elsharkawy, A., Thorne, A.W. and Mann, D.A. 2007. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell. Death. Differentiation. 14, 275–285; doi:10.1038/sj.cdd.4401979 Mannaerts, I., Leite, S.B., Verhulst, S., Claerhout, S., Eysackers, N., Thoen, L.F., Hoorens, A., Reynaert, H., Halder, G. and Van Grunsven, L.A. 2015. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 63, 679–688; doi:10.1016/j.jhep.2015.04.011 Marcher, A.B., Bendixen, S.M., Terkelsen, M.K., Hohmann, S.S., Hansen, M.H., Larsen, B.D., Mandrup, S., Dimke, H., Detlefsen, S. and Ravnskjaer, K. 2019. Transcriptional regulation of hepatic stellate cell activation in NASH. Sci. Rep. 9(1), 2324; doi: 10.1038/s41598-019-39112-6 Maretti-Mira, A.C., Wang, X., Wang, L. and Deleve, L.D. 2019. Incomplete differentiation of engrafted bone marrow endothelial progenitor cells initiates hepatic fibrosis in the rat. Hepatology 69, 1259–1272; doi:10.1002/hep.30227 Marra, F. and Tacke, F. 2014. Roles for chemokines in liver disease. Gastroenterology 147(1), 577–594; doi:10.1053/j.gastro.2014.06.043 Marra, F., Valente, A.J., Pinzani, M. and Abboud, H.E. 1993. Cultured human liver fat-storing cells produce monocyte chemotactic protein-1. Regulation by proinflammatory cytokines. J. Clin. Invest. 92, 1674–1680; doi:10.1172/JCI116753 Marra, F. 1999. Hepatic stellate cells and the regulation of liver inflammation. J. Hepatol. 31, 1120–1130; doi:10.1016/S0168-8278(99)80327-4 Masuya, M., Drake, C.J., Fleming, P.A., Reilly, C.M., Zeng, H., Hill, W.D., Martin-Studdard, A., Hess, D.C. and Ogawa, M. 2003. Hematopoietic origin of glomerular mesangial cells. Blood 101, 2215–2218; doi:10.1182/blood-2002-04-1076 Mazzocca, A., Dituri, F., Lupo, L., Quaranta, M., Antonaci, S. and Giannelli, G. 2011. Tumor-secreted lysophosphatidic acid accelerates hepatocellular carcinoma progression by promoting differentiation of peritumoral fibroblasts in myofibroblasts. Hepatology 54(3), 920–930; doi:10.1002/hep.24485 Mccommis, K.S., Hodges, W.T., Brunt, E.M., Nalbantoglu, I., Mcdonald, W.G., Holley, C., Fujiwara, H., Schaffer, J.E., Colca, J.R. and Finck, B.N. 2017. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 65, 1543–1556; doi:10.1002/hep.29025 Mederacke, I., Hsu, C.C., Troeger, J.S., Huebener, P., Mu, X., Dapito, D.H., Pradere, J.P. and Schwabe, R.F. 2013. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nature. Commun. 4(1), 1–10; doi:10.1038/ncomms3823 Mehal, W. 2023. Mechanisms of liver fibrosis in metabolic syndrome. eGastroenterology 1(1), e100015; doi:10.1136/egastro-2023-100015 Meurette, O. and Mehlen, P. 2018. Notch signaling in the tumor microenvironment. Cancer. Cell. 34, 536–548; doi:10.1016/j.ccell.2018.07.009 Miele, L., Valenza, V., La Torre, G., Montalto, M., Cammarota, G., Ricci, R., Mascianà, R., Forgione, A., Gabrieli, M.L., Perotti, G., Vecchio, F.M., Rapaccini, G., Gasbarrini, G., Day, C.P. and Grieco, A. 2009. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 49(6), 1877–1887; doi:10.1002/hep.22848 Miyahara, T., Schrum, L., Rippe, R., Xiong, S., Yee, H.F., Motomura, K., Anania, F.A., Willson, T.M. and Tsukamoto, H. 2000. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J. Biol. Chem. 275, 35715–35722; doi:10.1074/jbc.M006577200 Miyata, E., Masuya, M., Yoshida, S., Nakamura, S., Kato, K., Sugimoto, Y., Shibasaki, T., Yamamura, K., Ohishi, K., Nishii, K., Ishikawa, F., Shiku, H. and Katayama, N. 2008. Hematopoietic origin of hepatic stellate cells in the adult liver. Blood 111(4), 15; doi:10.1182/blood-2007-07-101261 Mooring, M., Fowl, B.H., Lum, S.Z.C., Liu, Y., Yao, K., Softic, S., Kirchner, R., Bernstein, A., Singhi, A.D., Jay, D.G., Kahn, C.R., Camargo, F.D. and Yimlamai, D. 2020. Hepatocyte Stress Increases Expression of Yes-Associated Protein and Transcriptional Coactivator with PDZ-Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology 71, 1813–1830; doi:10.1002/hep.30928 Moriwaki, H., Blaner, W.S., Piantedosi, R. and Goodman, D.S. 1988. Effects of dietary retinoid and triglyceride on the lipid composition of rat liver stellate cells and stellate cell lipid droplets. J. Lipid Res. 29, 1523–1534. Morrison, S.J., White, P.M., Zock, C. and Anderson, D.J. 1999. Prospective identification, isolation by flow cytometry, and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell 96, 737–749; doi:10.1016/S0092-8674(00)80583-8 Musso, G., Gambino, R. and Cassader, M. 2013. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 52, 175–191. doi: 10.1016/j.plipres.2012.11.002 Nasir, S.A., Mangla, A., Taneja, V., Berger, T., Pandya, D., Gupta, V. and Lim, J.K. 2025. Advances in Novel Drug Therapy for metabolic dysfunction-associated steatohepatitis cirrhosis. J. Transl. Gastroenterol. 3(1), 9–17; doi:10.14218/JTG.2024.00040 Neubauer, K., Knittel, T., Aurisch, S., Fellmer, P. and Ramadori, G. 1996. Glial fibrillary acidic protein—a cell type specific marker for Ito cells in vivo and in vitro. J. Hepatol. 24, 719–730; doi:10.1016/S0168-8278(96)80269-8 Ng, C.H., Lim, W.H., Hui Lim, G.E., Hao Tan, D.J., Syn, N., Muthiah, M.D., Huang, D.Q. and Loomba, R. 2023. Mortality outcomes by fibrosis stage in nonalcoholic fatty liver disease: a systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 21(4), 931–939; doi:10.1016/j.cgh.2022.04.014 Niki, T., Pekny, M., Hellemans, K., De Bleser, P., Van Den Berg, K., Vaeyens, F., Quartier, E., Schuit, F. and Geerts, A. 1999. Class VI intermediate filament protein nestin is induced during activation of rat hepatic stellate cells. Hepatology 29, 520–527; doi:10.1002/hep.510290232 O'Byrne, S.M., Wongsiriroj, N., Libien, J., Vogel, S., Goldberg, I.J., Baehr, W., Palczewski, K. and Blaner, W.S. 2005. Retinoid absorption and storage is impaired in mice lacking lecithin: retinol acyltransferase (LRAT). J. Biol. Chem. 280, 35647–35657; doi: 10.1074/jbc.M507924200 Ogawa, M., Larue, A.C. and Drake, C.J. 2006. Hematopoietic origin of fibroblasts/myofibroblasts: its pathophysiologic implications. Blood 108, 2893–2896; doi:10.1182/blood-2006-04-016600 Omenetti, A., Choi, S., Michelotti, G. and Diehl, A.M. 2011. Hedgehog signaling in the liver. J. Hepatol. 54, 366–373; doi:10.1016/j.jhep.2010.10.003 Otto, J., Verwaayen, A., Penners, C. and et al. 2023. Expression of Cyclin E1 in hepatic stellate cells is critical for the induction and progression of liver fibrosis and hepatocellular carcinoma in mice. Cell. Death. Discovery. 14, 549. Page, A., Paoli, P.P., Hill, S.J., Howarth, R., Wu, R., Kweon, S.M., French, J., White, S., Tsukamoto, H., Mann, D.A. and Mann, J. 2015. Alcohol directly stimulates epigenetic modifications in hepatic stellate cells. J. Hepatol. 62, 388–397; doi:10.1016/j.jhep.2014.09.033 Parola, M. and Pinzani, M. 2019. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 65, 37–55; doi:10.1016/j.mam.2018.09.002 Pellicoro, A., Ramachandran, P., Iredale, J.P. and Fallowfield, J.A. 2014. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 14, 181–194; doi:10.1038/nri3623 Peters, K.M., Dommaschk, J., Grundmann, R., Schaadt, M. and Schicha, H. 1990. Monitoring of tumor necrosis factor therapy with neopterin. Arzneimittelforschung 40, 508–510. Pinzani, M., Abboud, H.E., Gesualdo, L. and Abboud, S.L. 1992. Regulation of macrophage colony-stimulating factor in liver fat storing cells by peptide growth factors. Am. J. Physiol. 262, C876–C881; doi:10.1152/ajpcell.1992.262.4.C876 Pinzani, M., Carloni, V., Marra, F., Riccardi, D., Laffi, G. and Gentilini, P. 1994. Biosynthesis of platelet-activating factor and its 1O-acyl analogue by liver fat-storing cells. Gastroenterology 106, 1301–1311; doi:10.1016/0016-5085(94)90023-x Potter, J.J., Rennie-Tankesley, L. and Mezey, E. 2003. Influence of leptin in the development of hepatic fibrosis produced in mice by Schistosoma mansoni infection and by chronic carbon tetrachloride administration. J. Hepatol. 38(3), 281–288; doi:10.1016/s0168-8278(02)00414-2 Pradere, J.P., Kluwe, J., De Minicis, S., Jiao, J.J., Gwak, G.Y., Dapito, D.H., Jang, M.K., Guenther, N.D., Mederacke, I., Friedman, R., Dragomir, A.C., Aloman, C. and Schwabe, R.F. 2013. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473; doi:10.1002/hep.26429 Ramm, G.A. 2009. Chemokine (C-C motif) receptors in fibrogenesis and hepatic regeneration following acute and chronic liver disease. Hepatology 50, 1664–1668; doi:10.1002/hep.23338 Rockey, D.C. and Chung, J.J. 1994. Interferon gamma inhibits lipocyte activation and extracellular matrix mRNA expression during experimental liver injury: implications for treatment of hepatic fibrosis. J. Investig. Med. 42, 660–670; doi:10.1136/jim-42-4-660 Roh, Y.J., Kim, H. and Choi, D.W. 2025. Metabolic Sparks in the Liver: metabolic and Epigenetic Reprogramming in Hepatic Stellate Cells Activation and Its Implications for Human Metabolic Diseases. Diabetes. Metab. J. 49(3), 368–385; doi:10.4093/dmj.2025.0195 Rossi, D. and Zlotnik, A. 2000. The biology of chemokines and their receptors. Annu. Rev. Immunol. 18, 217–242; doi:10.1146/annurev.immunol.18.1.217 Russo, F.P., Alison, M.R., Bigger, B.W., Amofah, E., Florou, A., Amin, F., Bou–Gharios, G., Jeffery, R., Iredale, J.P. and Forbes, S.J. 2006. The bone marrow functionally contributes to liver fibrosis. Gastroenterology 130, 1807–1821; doi:10.1053/j.gastro.2006.01.036 Sahin, H., Borkham-Kamphorst, E., Kuppe, C., Zaldivar, M.M., Grouls, C., Al-Samman, M., Nellen, A., Schmitz, P., Heinrichs, D., Berres, M.L., Doleschel, D., Scholten, D., Weiskirchen, R., Moeller, M.J., Kiessling, F., Trautwein, C. and Wasmuth, H.E. 2012. Chemokine Cxcl9 attenuates liver fibrosis associated angiogenesis in mice. Hepatology 55, 1610–1619; doi:10.1002/hep.25545 Saiman, Y. and Friedman, S.L. 2012. The role of chemokines in acute liver injury. Front. Physiol. 3, 213; doi:10.3389/fphys.2012.00213 Sato, M., Suzuki, S. and Senoo, H. 2003. Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell. Struct. Funct. 28(2), 105–112; doi:10.1247/csf.28.105 Sawitza, I., Kordes, C., Reister, S. and Häussinger, D. 2009. The niche of stellate cells within rat liver. Hepatology 50, 1617–1624; doi:10.1002/hep.23184 Schmitt-Gräff, A., Chakroun, G. and Gabbiani, G. 1993. Modulation of perisinusoidal cell cytoskeletal features during experimental hepatic fibrosis. Virchows. Arch. A. Pathol. Anat. Histopathol. 422, 99–107; doi:10.1007/BF01607166 Schunk, S.J., Floege, J., Fliser, D. and Speer, T. 2021. WNT-β-catenin signalling—A versatile player in kidney injury and repair. Nat. Rev. Nephrol. 17, 172–184; doi:10.1038/s41581-020-00343-w Schuppan, D. and Kim, Y.O. 2013. Evolving therapies for liver fibrosis. J. Clin. Invest. 123(5), 1887–1901; doi: 10.1172/JCI66028 Schuster, S., Cabrera, D., Arrese, M. and Feldstein, A.E. 2018. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 15(6), 349–364; doi:10.1038/s41575-018-0009-6 Schwabe, R.F., Bataller, R. and Brenner, D.A. 2003. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G949–G958; doi:10.1152/ajpgi.00215.2003 Schwabe, R.F., Tabas, I. and Pajvani, U.B. 2020. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158, 1913–1928; doi:10.1053/j.gastro.2019.11.311 Seki, E. and Brenner, D.A. 2015. Recent advancement of molecular mechanisms of liver fibrosis. J. Hepatobiliary. Pancreat. Sci. 22, 512–518; doi:10.1002/jhbp.245 Seki, E. and Schwabe, R.F. 2015. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 61(3), 1066–1079; doi:10.1002/hep.27332 Seki, E., De Minicis, S., Gwak, G.Y., Kluwe, J., Inokuchi, S., Bursill, C.A., Llovet, J.M., Brenner, D.A. and Schwabe, R.F. 2009. CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Invest. 119, 1858–1870; doi:10.1172/JCI37444 Seki, E., De Minicis, S., Osterreicher, C.H., Kluwe, J., Osawa, Y., Brenner, D.A. and Schwabe, R.F. 2007. TLR4 Enhances TGF-beta Signaling and Hepatic Fibrosis. Nat. Med. 13, 1324–1332; doi:10.1038/nm1663 Semba, R., Tanaka, O. and Tanimura, T. 1983. Digestive System.In Atlas of Human Prenatal Histology. Nishimura H (ed) Tokyo, Japan: Igaku-shoin, pp: 171–240. Senoo, H. 2004. Structure and function of hepatic stellate cells. Med. Electron. Microsc. 37, 3–15; doi:10.1007/s00795-003-0230-3 Senoo, H., Hata, R., Nagai, Y. and Wake, K. 1984. Stellate cells (vitamin A-storing cells) are the primary site of collagen synthesis in nonparenchymal cells in the liver. Biomed. Res. 5, 451–458. Senoo, H., Yoshikawa, K., Morii, M., Miura, M., Imai, K. and Mezaki, Y. 2010. Hepatic stellate cell (vitamin A-storing cell) and its relative—past, present and future. Cell. Biol. Int. 34(12), 1247–1272; doi:10.1042/CBI20100321 Sererols-Viñas, L., Garcia-Vicién, G., Ruiz-Blázquez, P., Lee, T.F., Lee, Y.A., Gonzalez-Sanchez, E., Vaquero, J., Moles, A., Filliol, A. and Affò, S. 2025. Hepatic Stellate Cells Functional Heterogeneity in Liver Cancer. Semin. Liver Dis. 45(1), 33–51; doi:10.1055/a-2551-0724 Shackel, N.A. 2001. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut 49, 565–576; doi:10.1136/gut.49.4.565 She, H., Xiong, S., Hazra, S. and Tsukamoto, H. 2005. Adipogenic transcriptional regulation of hepatic stellate cells. J. Biol. Chem. 280, 4959–4967; doi:10.1074/jbc.M410078200 Shi, M., Zhu, J., Wang, R., Chen, X., Mi, L., Walz, T. and Springer, T.A. 2011. Latent TGF-β structure and activation. Nature 474, 343–349; doi:10.1038/nature10152 Shmarakov, I.O., Jiang, H., Liu, J., Fernandez, E.J. and Blaner, W.S. 2019. Hepatic stellate cell activation: a source for bioactive lipids. Biochimica. Et. Biophysica. Acta. -. Mol. Cell. Biol. Lipids. 1864, 629–642; doi:10.1016/j.bbalip.2019.02.004 Sica, A., Invernizzi, P. and Mantovani, A. 2014. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 59, 2034–2042; doi:10.1002/hep.26754 Slack, R.J., Mills, R. and Mackinnon, A.C. 2021. The therapeutic potential of galectin-3 inhibition in fibrotic disease. Int. J. Biochem. Cell Biol. 130, 105881; doi:10.1016/j.biocel.2020.105881 Subramanian, P., Hampe, J., Tacke, F. and Chavakis, T. 2022. Fibrogenic pathways in metabolic dysfunction associated fatty liver disease (MAFLD). Int. J. Mol. Sci. 23, 6996; doi:10.3390/ijms23136996 Tacke, F., Zimmermann, H.W., Berres, M.L., Trautwein, C. and Wasmuth, H.E. 2011. Serum chemokine receptor CXCR3 ligands are associated with progression, organ dysfunction and complications of chronic liver diseases. Liver. Int. 31, 840–849; doi:10.1111/j.1478-3231.2011.02504.x Tao, L., Wu, L., Zhang, W., Ma, W.T., Yang, G.Y., Zhang, J., Xue, D.Y., Chen, B. and Liu, C. 2020. Peroxisome proliferator-activated receptor gamma inhibits hepatic stellate cell activation regulated by miR-942 in chronic hepatitis B liver fibrosis. Life Sci. 253, 117572; doi:10.1016/j.lfs.2020.117572 Taschler, U., Schreiber, R., Chitraju, C., Grabner, G.F., Romauch, M., Wolinski, H., Haemmerle, G., Breinbauer, R., Zechner, R., Lass, A. and Zimmermann, R. 2015. Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells. Biochim. Biophys. Acta. 1851, 937–945; doi:10.1016/j.bbalip.2015.02.017 Taylor, R.S., Taylor, R.J., Bayliss, S., Hagström, H., Nasr, P., Schattenberg, J.M., Ishigami, M., Toyoda, H., Wai-sun Wong, V., Peleg, N., Shlomai, A., Sebastiani, G., Seko, Y., Bhala, N., Younossi, Z.M., Anstee, Q.M., Mcpherson, S. and Newsome, P.N. 2020. Association between fibrosis stage and outcomes of patients with nonalcoholic fatty liver disease: a systematic review and meta-analysis. Gastroenterology 158, 1611–1625; doi:10.1053/j.gastro.2020.01.043 Thakral, S. and Ghoshal, K. 2015. MiR-122 Is a Unique Molecule with Great Potential in Diagnosis, Prognosis of Liver Disease, and Therapy Both as miRNA Mimic and Antimir. Curr. Gene. Ther. 15(2), 142–150. Tian, X., Wang, Y., Lu, Y., Wang, W., Du, J., Chen, S., Zhou, H., Cai, W. and Xiao, Y. 2021. Conditional depletion of macrophages ameliorates cholestatic liver injury and fibrosis via lncRNA-H19. Cell Death Dis. 12, 646; doi:10.1038/s41419-021-03931-1 Tiggelman, A.M., Boers, W., Linthorst, C., Sala, M. and Chamuleau, R.A. 1995. Collagen synthesis by human liver (myo)fibroblasts in culture: evidence for a regulatory role of IL-1 beta, IL-4, TGF beta and IFN gamma. J. Hepatol. 23, 307–317; doi:10.1016/S0168-8278(95)80010-7 Trautwein, C., Friedman, S.L., Schuppan, D. and Pinzani, M. 2015. Hepatic fibrosis: concept to treatment. J. Hepatol. 62(1), S15–S24; doi:10.1016/j.jhep.2015.02.039 Trivedi, P., Wang, S. and Friedman, S.L. 2021. The Power of Plasticity—Metabolic Regulation of Hepatic Stellate Cells. Cell Metab. 33, 242–257; doi:10.1016/j.cmet.2020.10.026 Tsuchida, T. and Friedman, S.L. 2017. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14(7), 397–411; doi:10.1038/nrgastro.2017.38 Tsukamoto, H. 2005. Adipogenic phenotype of hepatic stellate cells. Alcohol. Clin. Exp. Res. 29, 132S–133S; doi:10.1097/01.alc.0000189279.92602.f0 Wake, K. 1980. Perisinusoidal stellate cells (fat-storing cells, interstitial cells, lipocytes), their related structure in and around the liver sinusoids, and vitamin A-storing cells in extrahepatic organs. Int. Rev. Cytol. 66, 303–353. doi: 10.1016/S0074-7696(08)61977-4 Wake, K., Motomatsu, K. and Ekataksin, W. 1991. Postnatal development of the perisinusoidal stellate cells in thre rat liver.In Cells of the hepatic sinusoid. Wisse D Knook., McCuskey. and Eds. Rijswijk, The Netherlands: Kupffer Cell Foundation, pp: 269–75. Wake, K. 1995. Structure of the sinusoidal wall in the liver. In Cells of the Hepatic Sinusoid Eds., Wisse, D.L. and Wake, K.. Leiden, The Netherlands: The Kupffer Cell Foundation, pp: 241–6. Wake, K. 1999. Cell-cell organization and functions of 'sinusoids' in liver microcirculation system. J. Electron Microsc. (Tokyo) 48, 89–98. doi: 10.1093/oxfordjournals.jmicro.a023666 Wang, L., Tankersley, L.R., Tang, M., Potter, J.J. and Mezey, E. 2002. Regulation of the murine alpha(2)(I) collagen promoter by retinoic acid and retinoid X receptors. Arch. Biochem. Biophys. 401, 262–270; doi:10.1016/S0003-9861(02)00058-9 Wang, M., Li, L., Xu, Y., Du, J. and Ling, C. 2022. Roles of hepatic stellate cells in NAFLD: from the perspective of inflammation and fibrosis. Front. Pharmacol. 13, 958428; doi:10.3389/fphar.2022.958428 Wang, Q., Wei, S., Li, L., Bu, Q., Zhou, H., Su, W., Liu, Z., Wang, M. and Lu, L. 2021. MiR-139-5p sponged by LncRNA NEAT1 regulates liver fibrosis via targeting beta-catenin/SOX9/TGF-beta1 pathway. Cell Death Discovery 7, 243; doi:10.1038/s41420-021-00632-8 Wang, S.C., Ohata, M., Schrum, L., Rippe, R.A. and Tsukamoto, H. 1998. Expression of interleukin-10 by in vitro and in vivo activated hepatic stellate cells. J. Biol. Chem. 273(1), 302–308; doi:10.1074/jbc.273.1.302 Wells, R. and Schwabe, R. 2015. Origin and Function of Myofibroblasts in the Liver. Semin. Liver. Dis. 35(2), 97–106. Wiering, L., Subramanian, P. and Hammerich, L. 2023. Hepatic Stellate Cells: dictating Outcome in Nonalcoholic Fatty Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 15, 1277–1292; doi:10.1016/j.jcmgh.2023.02.010 Winwood, P. 1995. Kupffer cell-derived 95-kd type IV collagenase/gelatinase B: characterization and expression in cultured cells. Hepatology 22, 304–315. Wree, A., Eguchi, A., Mcgeough, M.D., Pena, C.A., Johnson, C.D., Canbay, A., Hoffman, H.M. and Feldstein, A.E. 2014. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 59, 898–910; doi:10.1002/hep.26592 Xie, G., Wang, X., Wang, L., Wang, L., Atkinson, R.D., Kanel, G.C., Gaarde, W.A. and Deleve, L.D. 2012. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 142, 918–927; doi:10.1053/j.gastro.2011.12.017 Xu, J.J., Chen, X., Zhu, S., Jiang, L.F., Ma, W.X., Chen, S.Y., Meng, X.M., Huang, C. and Li, J. 2023. Myc-mediated circular RNA circMcph1/miR-370-3p/Irak2 axis is a progressive regulator in hepatic fibrosis. Life Sci. 312, 121182; doi:10.1016/j.lfs.2022.121182 Yan, N. 2017. A glimpse of membrane transport through structures—advances in the structural biology of the GLUT glucose transporters. J. Mol. Biol. 429, 2710–2725. Yin, K.L., Li, M., Song, P.P., Duan, Y.X., Ye, W.T., Tang, W., Kokudo, N., Gao, Q. and Liao, R. 2023. Unraveling the emerging niche role of hepatic stellate cell-derived exosomes in liver diseases. J. Clin. Transl. Hepatol. 11, 441–451; doi:10.14218/JCTH.2022.00326 Yoneda, A., Sakai-Sawada, K., Niitsu, Y. and Tamura, Y. 2016. Vitamin A and insulin are required for the maintenance of hepatic stellate cell quiescence. Exp. Cell. Res. 341(8), 9–17; doi:10.1016/j.yexcr.2016.01.012 You, K., Li, S.Y., Gong, J., Fang, J.H., Zhang, C., Zhang, M., Yuan, Y., Yang, J. and Zhuang, S.M. 2018. MicroRNA-125b Promotes Hepatic Stellate Cell Activation and Liver Fibrosis by Activating RhoA Signaling. Mol. Therapy. Nucleic Acids 12, 57–66; doi:10.1016/j.omtn.2018.04.016 Yu, Q. and Stamenkovic, I. 2000. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes. Dev. 14, 163–176; doi:10.1101/gad.14.2.163 Zheng, H., You, Y., Hua, M., Wu, P., Liu, Y., Chen, Z., Zhang, L., Wei, H., Li, Y., Luo, M., Zeng, Y., Liu, Y., Luo, D.X., Zhang, J., Feng, M., Hu, R., Pandol, S.J. and Han, Y.P. 2018. Chlorophyllin Modulates Gut Microbiota and Inhibits Intestinal Inflammation to Ameliorate Hepatic Fibrosis in Mice. Front. Physiol. 9, 1671; doi:10.3389/fphys.2018.01671 | ||
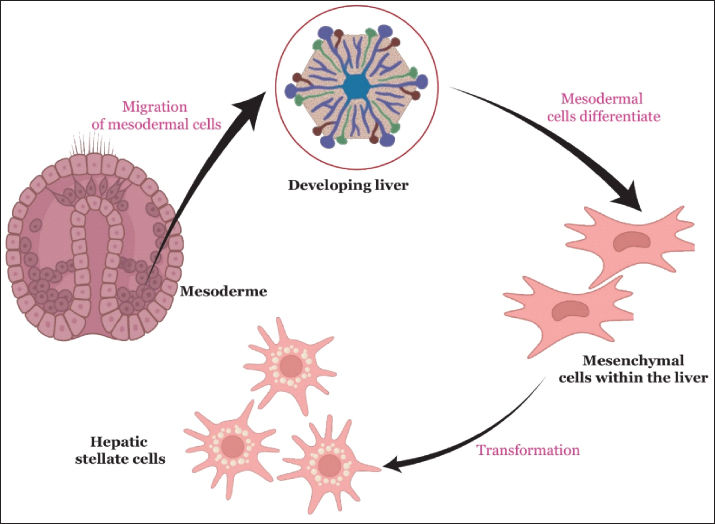
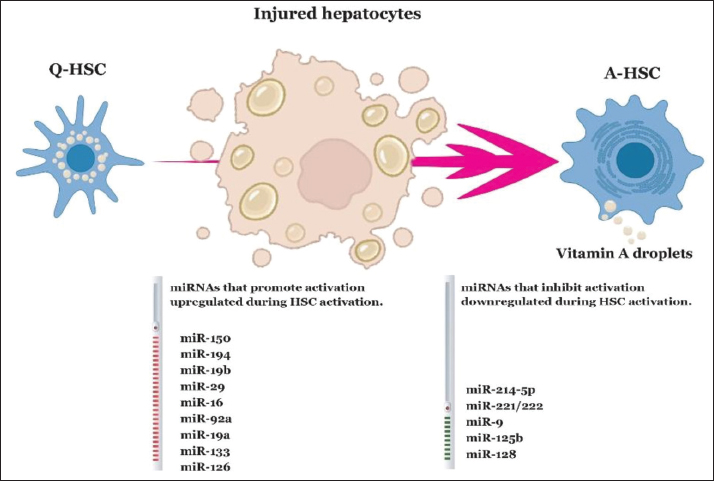
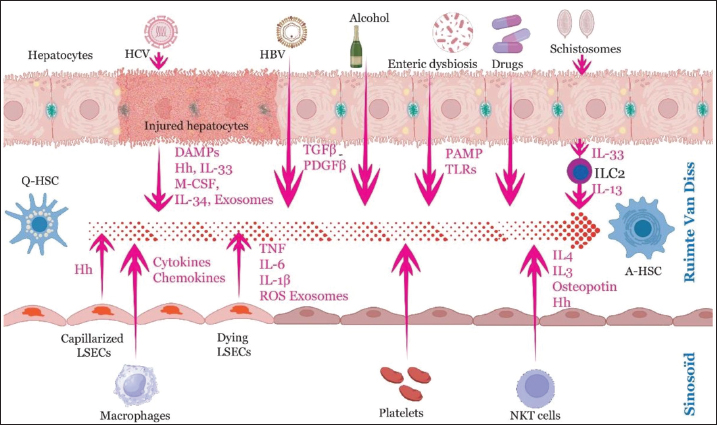
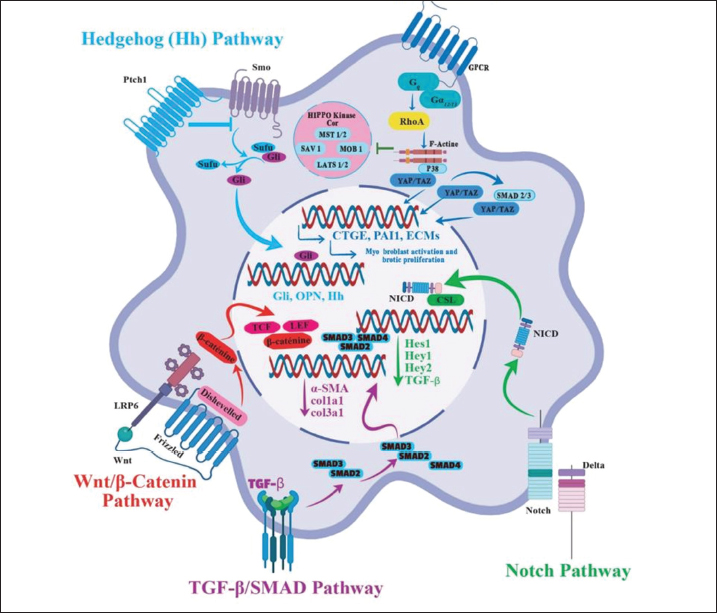
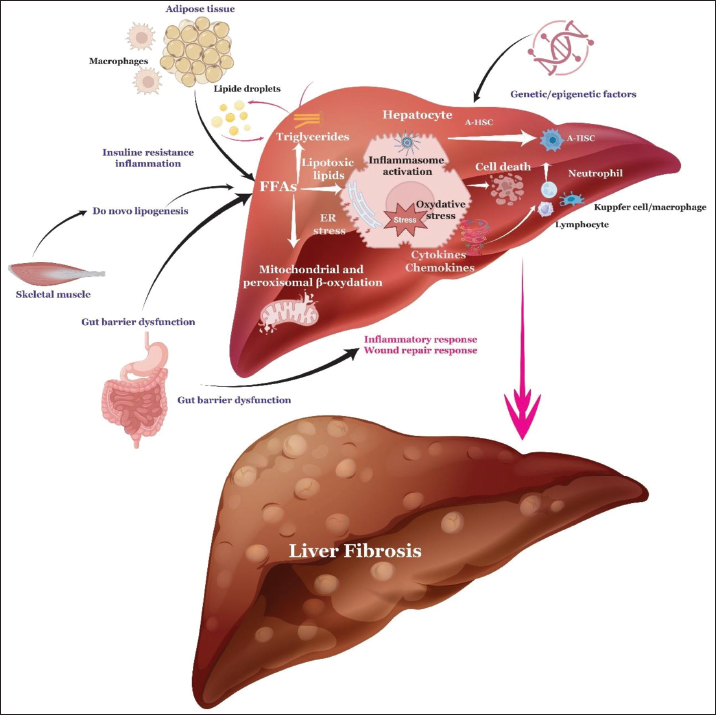

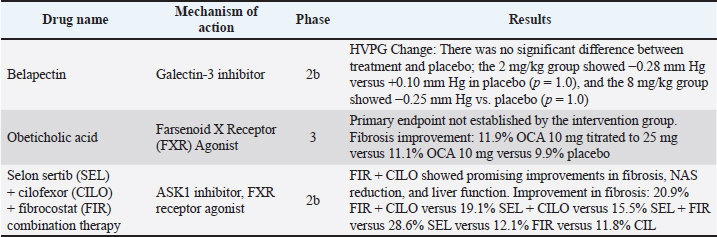
| How to Cite this Article |
| Pubmed Style Esselmani H, Nadir Y, Ennadi R, Zouhairi N, Hiba OE, Lkousse MA, Najimi M, Merzouki M. Hepatic stellate cells in liver fibrosis: From biology to pathology. Open Vet. J.. 2025; 15(12): 6190-6214. doi:10.5455/OVJ.2025.v15.i12.4 Web Style Esselmani H, Nadir Y, Ennadi R, Zouhairi N, Hiba OE, Lkousse MA, Najimi M, Merzouki M. Hepatic stellate cells in liver fibrosis: From biology to pathology. https://www.openveterinaryjournal.com/?mno=276849 [Access: June 26, 2026]. doi:10.5455/OVJ.2025.v15.i12.4 AMA (American Medical Association) Style Esselmani H, Nadir Y, Ennadi R, Zouhairi N, Hiba OE, Lkousse MA, Najimi M, Merzouki M. Hepatic stellate cells in liver fibrosis: From biology to pathology. Open Vet. J.. 2025; 15(12): 6190-6214. doi:10.5455/OVJ.2025.v15.i12.4 Vancouver/ICMJE Style Esselmani H, Nadir Y, Ennadi R, Zouhairi N, Hiba OE, Lkousse MA, Najimi M, Merzouki M. Hepatic stellate cells in liver fibrosis: From biology to pathology. Open Vet. J.. (2025), [cited June 26, 2026]; 15(12): 6190-6214. doi:10.5455/OVJ.2025.v15.i12.4 Harvard Style Esselmani, H., Nadir, . Y., Ennadi, . R., Zouhairi, . N., Hiba, . O. E., Lkousse, . M. A., Najimi, . M. & Merzouki, . M. (2025) Hepatic stellate cells in liver fibrosis: From biology to pathology. Open Vet. J., 15 (12), 6190-6214. doi:10.5455/OVJ.2025.v15.i12.4 Turabian Style Esselmani, Hicham, Youssef Nadir, Rahma Ennadi, Nadia Zouhairi, Omar El Hiba, Mohammed Amine Lkousse, Mustapha Najimi, and Mohamed Merzouki. 2025. Hepatic stellate cells in liver fibrosis: From biology to pathology. Open Veterinary Journal, 15 (12), 6190-6214. doi:10.5455/OVJ.2025.v15.i12.4 Chicago Style Esselmani, Hicham, Youssef Nadir, Rahma Ennadi, Nadia Zouhairi, Omar El Hiba, Mohammed Amine Lkousse, Mustapha Najimi, and Mohamed Merzouki. "Hepatic stellate cells in liver fibrosis: From biology to pathology." Open Veterinary Journal 15 (2025), 6190-6214. doi:10.5455/OVJ.2025.v15.i12.4 MLA (The Modern Language Association) Style Esselmani, Hicham, Youssef Nadir, Rahma Ennadi, Nadia Zouhairi, Omar El Hiba, Mohammed Amine Lkousse, Mustapha Najimi, and Mohamed Merzouki. "Hepatic stellate cells in liver fibrosis: From biology to pathology." Open Veterinary Journal 15.12 (2025), 6190-6214. Print. doi:10.5455/OVJ.2025.v15.i12.4 APA (American Psychological Association) Style Esselmani, H., Nadir, . Y., Ennadi, . R., Zouhairi, . N., Hiba, . O. E., Lkousse, . M. A., Najimi, . M. & Merzouki, . M. (2025) Hepatic stellate cells in liver fibrosis: From biology to pathology. Open Veterinary Journal, 15 (12), 6190-6214. doi:10.5455/OVJ.2025.v15.i12.4 |

