




| Original Article | ||
Open Vet J. 2020; 10(1): 94-115 doi: 10.4314/ovj.v10i1.15 Open Veterinary Journal, (2020), Vol. 10(1): 94–115 Original Research DOI: http://dx.doi.org/10.4314/ovj.v10i1.15 Genomic characteristics of classical swine fever virus strains of bovine origin according to primary and secondary sequence–structure analysisMassimo Giangaspero1* and Shu-Qin Zhang21Faculty of Veterinary Medicine, University of Teramo, Italy 2State Key Laboratory for Molecular Biology of Special Economic Animals, Institute of Special Economic Animal and Plant Sciences, No 4899 Juye Street, Changchun, Jilin, People’s Republic of China *Corresponding Author: Massimo Giangaspero. Faculty of Veterinary Medicine, University of Teramo, Italy. Email: giangasp [at] gmail.com Submitted: 29/12/2019 Accepted: 04/03/2020 Published: 27/03/2020 © 2020 Open Veterinary Journal
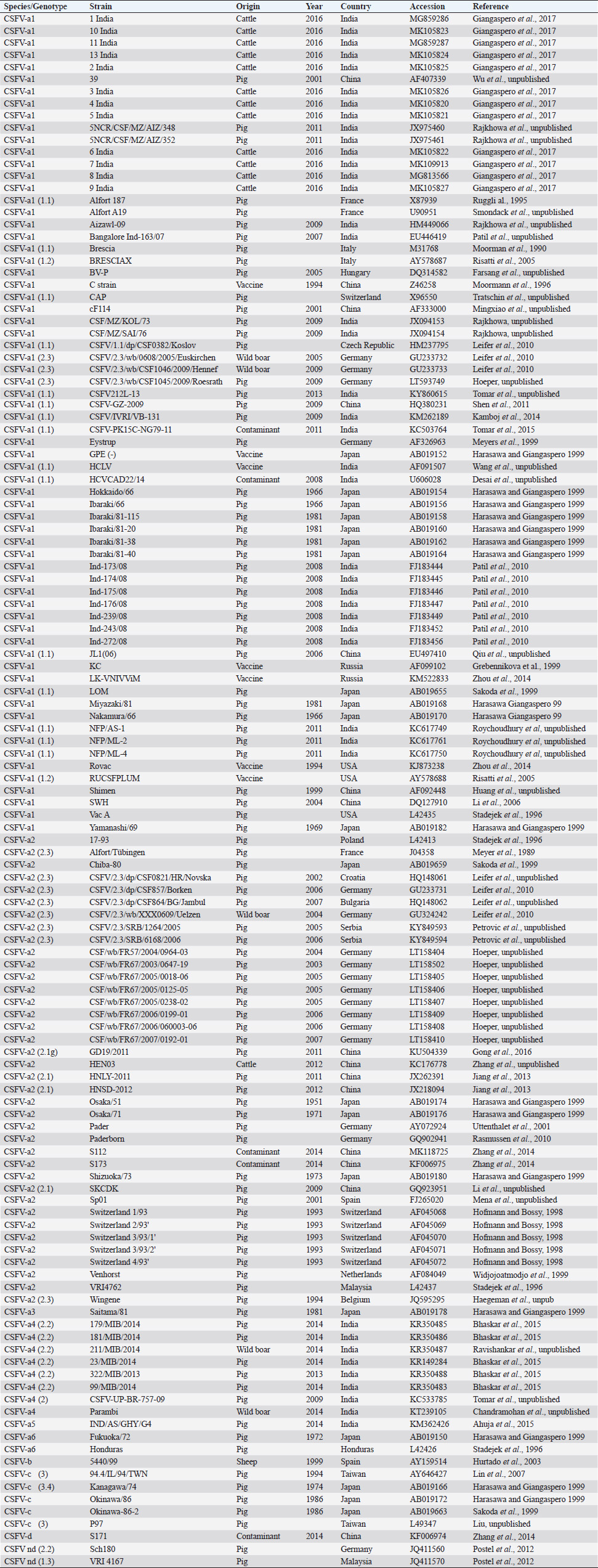
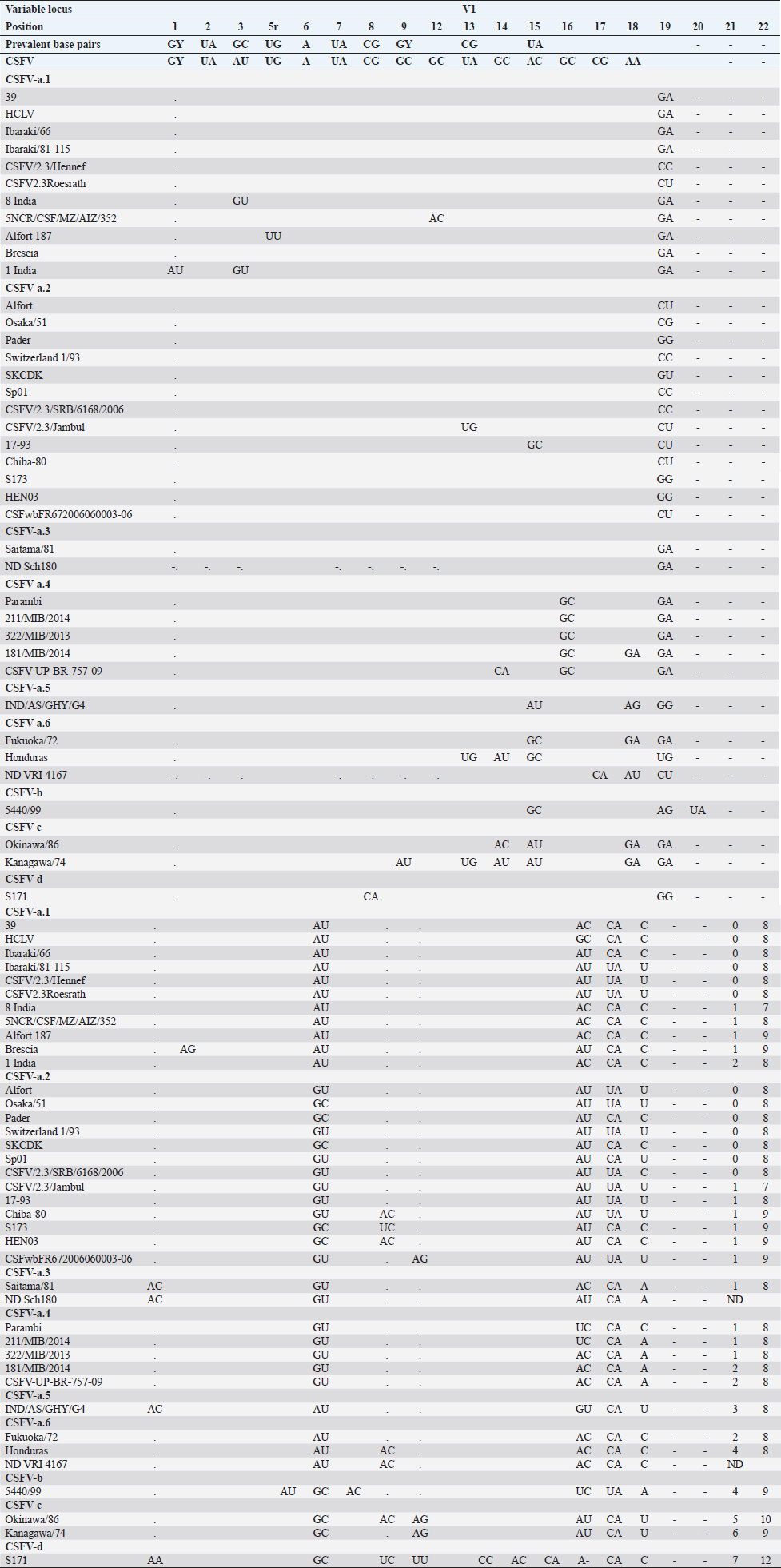
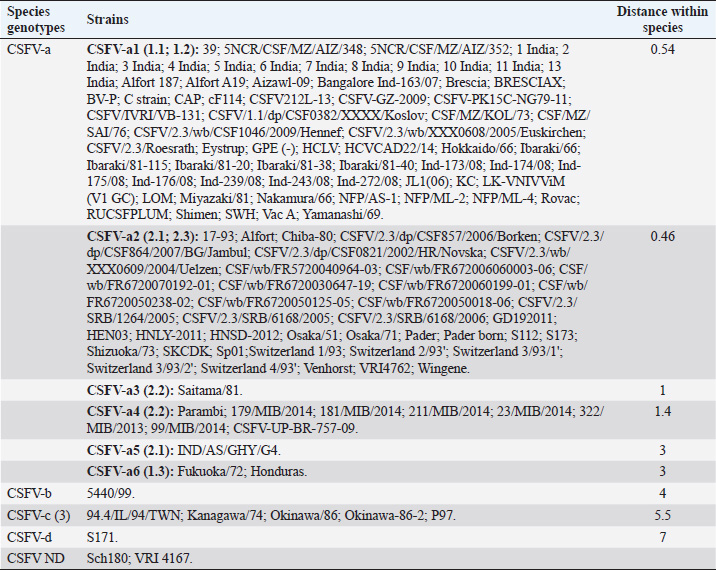
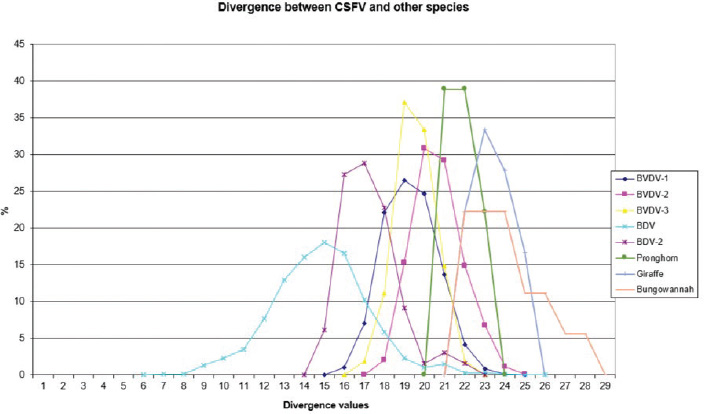
AbstractBackground: Classical swine fever virus (CSFV), species member of the family Flaviviridae, is generally considered restricted to domestic and wild suids. The circulation of CFSV has been detected in cattle herds in China and India. Natural infection appeared associated with clinical signs in some cases. Aim: The secondary structures of the internal ribosome entry site in the 5′ untranslated region (UTR) were used for the genomic characterization of bovine strains. Methods: Sequences have been compared to the representative CSFV strains isolated from pigs, vaccines, and contaminants from porcine cell lines and an ovine strain isolated in Spain. Results: The observed sequences from cattle showed a genetic relatedness with live-attenuated vaccine strains used in pigs. Sequence characteristics of the Chinese strain S171 are genetically distant from the previously reported CSFV genotypes, suggesting a new outgroup in the species, described for the first time, and named CSFV-d. Other Chinese strains were genetically closely related to CSFV genotype a2 (Alfort type) pig strains. Indian strains, reported from the states of Tamil Nadu and Meghalaya, were genetically closely related to CSFV genotype a1 (Brescia type) and a5 pig strains, respectively. Conclusion: These preliminary observations are new and relevant in countries, where CSFV control and eradication strategies are applied. Keywords: Cattle, Classical swine fever virus, Pestivirus, Secondary structure. IntroductionThe genus Pestivirus, family Flaviviridae, includes genetically related species recognized as important pathogens in veterinary medicine. The Classical swine fever virus (CSFV) has been eradicated in different countries as in Europe and North America, but still control and eradication strategies are applied against the virus in various other regions. There is a consensus concerning the capacity among pestiviruses to cross species barrier. The wide host range of these viruses may lead to the formation of virus reservoirs in other domestic or wildlife species, presenting a concern for the long-term success of eradication campaigns. Nevertheless, CSFV is generally considered to be restricted to domestic and wild suids. Current knowledge indicates that experimental infections without clinical signs have been reported in cattle, sheep, goats, and deer, but there is no evidence that these species become infected in nature (Biró et al., 1966; Loan and Storm, 1968; Shimizu and Kumagai, 1989; CFSPH, 2015). In only two cases, the species has been identified in sheep (Hurtado et al., 2003; Mohamed, 2004), corroborating these beliefs. One ovine isolate (strain 5440/99), which is similar to CSFV vaccine strains used in the framework of eradication campaigns in pigs, was reported from Spain (Hurtado et al., 2003). A second isolate (strain 12 Ovine liver 113nt) was obtained during investigations on pestiviruses in the United Arab Emirates (Mohamed, 2004). This Pestivirus-like sequence, when aligned with corresponding sequences from a variety of pestiviruses, showed a greatest similarity to CSFV subgenotype 1.1 but raised doubts on correct classification, due to the suboptimal quality of the sequence and its short length (113 nucleotides) (Mohamed, 2004). On contrary to this apparent epidemiological static nature and apparent inability to cross species barrier in natural conditions, the recent reports indicated the isolation of CSFV strains in cattle (Bos taurus) from India and China and contaminants or associated to natural infection with reproductive disorders (abortion, stillbirth, birth defects, repeat breeding, and mastitis) (Zhang et al., 2014; Giangaspero et al., 2017). Twelve strains isolated from cattle aged from 6 months to 2 years have been detected in four farms, out of 19 tested, during an epidemiological survey aiming to identify bovine viral diarrhea virus (BVDV) persistently infected animals by reverse transcription-polymerase chain reaction (RT-PCR), among BVDV-seropositive enzyme-linked immunosorbent assay (ELISA) cattle and buffalo farms from four districts of the state of Tamil Nadu, India (Giangaspero et al., 2017). Four strains were reported from China. Pestivirus RNA sequences have been detected from commercial fetal bovine serum batches and designated as strains S171 (KF006974), S173 (KF006975), and S112 (MK118725) (identical to S173) originated from Henan and Jiangsu provinces (Zhang et al., 2014). The attempts to isolate live viruses from some of these samples have been unsuccessful. The strain HEN03 (sequence deposited by Zhang, under accession number KC176778) was detected in cattle from Henan province. In addition, during a survey for the detection and genetic characterization of 5′-untranslated region (UTR) and E2 gene of CSFV from bovine population of the northeastern region of the Indian state of Meghalaya, of 134 cattle serum samples tested, all were positive in RT-PCR for 5′-UTR region, and 10 samples were positive for CSFV antigen by a commercial antigen capture ELISA (Chakraborty et al., 2018). The full-length E2 region of CSFV was amplified from two positive samples. In this study, the 5′-UTR sequences of the reported strains have been considered for genomic characterization according to the secondary structure analysis of the internal ribosome entry site (IRES). Material and MethodsThe 5′-UTR sequences of the 16 reported strains (13 sequences detected in cattle from India and China and three sequences detected in commercial fetal bovine serum batches from China) (Zhang et al., 2014; Giangaspero et al., 2017; and with reference to strain HEN03 sequence deposited by Zhang) have been compared with those from other CSFV strains (n=110) isolated from pigs, vaccines, and contaminants from porcine cell lines and an ovine strain isolated in Spain representative of identified genetic clusters in the species, with particular attention to isolates circulating in Asian countries, including India and China (Table 1), and further compared to 1,300 sequences from other Pestivirus species different from CSFV. The two Indian bovine CSFV sequences reported by Chakraborty et al. (2018) were evaluated indirectly considering the similarity of their full-length E2 region with other previously deposited pig sequences. Newly described subgenotypes reported from Brazil and Cuba (Postel et al., 2013; Silva et al., 2017) could not be considered, being available only E2 sequences, and apparently specific and restricted to South American and Caribbean regions. The taxonomical segregation of the genomic sequences was performed according to the genotyping based on the palindromic nucleotide substitution (PNS) method (Harasawa and Giangaspero, 1998; Giangaspero and Harasawa, 2007; Giangaspero and Apicella, 2014), through the qualitative and quantitative evaluation of relevant secondary structure regions from full-length (216–237 nt) 5′-UTR of the viral RNA, the three variable regions, and V1, V2, and V3 genomic sequences in the IRES. Genotypes were identified according to base–pair (bp) combinations at the level of low-variable positions and ranked with alphabetic nomenclature according to increasing divergence in the species. The classification among CSFV strains according to the PNS analysis based on changes in the secondary structure was compared with those based on the primary structure of the 5′-UTR performed through sequence alignment and construction of phylogenetic trees. A phylogenetic tree was constructed with Clustal X (Chenna et al., 2003) by using the neighbor-joining method (Saitou and Nei, 1987) and visualized with the Newick tree format option (Mega version 7.0.26) (Kumar et al., 2016). In addition, a basic local alignment search tool (BLAST; http://www.ncbi.nlm.nih.gov), which is the web-based sequence analysis tool with default values, was used to find the homologous hits for the sequence analysis of strains. ResultsThe obtained secondary structures have been aligned (Table 2) for computing genetic distance among sequences, in terms of base-pairing variations at the level of the IRES in the 5′-UTR, to identify genetic groups, ordered according to the increasing values of divergence. Among Indian bovine CSFV sequences, the V2 and V3 loci are all identical. The sequence of 8 India is identical to the sequences 2, 3, 9, and 13. The sequence of 1 India was identical to 4, 5, 6, 7, 10, and 11 (11 India showed only the first nucleotide in V1 locus: adenine instead of guanine). CSFV-c strain Okinawa/86 is identical to the strains P97 and 94.4/IL/94/TWN (difference in V1/1 g-c), CSFV-a1 strain Ibaraki/66 was identical to BresciaX (difference in V2/7 g*u), and CSFV-a1 39 was identical to strains JL1(06), 5NCRCSFMZAIZ348, Aizawl-09, and RUCSFPLUM (difference in V1/1 g-c). The sequences have been clustered into four genotypes, from CSFV-a to CSFV-d (Table 3). PNS marker characteristics to the Pestivirus CSFV species, genotype, and subgenotypes are shown in Table 4. The secondary sequence analysis of the 5’-UTR sequences of the Chinese and Indian strains suggested their appurtenance to CSFV species, genotype CSFV-a2 (Alfort type) for three of the Chinese strains and genotype CSFV-a1 (Brescia type) for all the Indian strains. The Chinese strain S171 was clustered as a new genotype in the species, named CSFV-d. Only partially related to CSFV-a2 (divergence 62.5%, mean value 9.87), with a marked difference at the level of the V3 locus nucleotide base pairings, S171 was divergent from all other CSFV genotypes (divergence values ranging from 10 to 15). Furthermore, it was significantly less related to border disease virus (BDV) (Fig. 1), a known characteristic of other CSFV strains. Base pairing comparison between strain S171 and strains of the different BDV genotypes scored the high divergence values, ranging from 17 to 25, with a mean value of 20.31. These values were very high in confront of the mean divergence value of 14.57 obtained compared to the other CSFV strains with BDV strains (Table 5). Divergence from BDV-2 tentative species is also higher with CSFV-d (divergence values ranging from 21 to 22; mean divergence value 21.33), in confront of scores obtained with other CSFV genotypes CSFV-a, CSFV-b, and CSFV-c (divergence values ranging from 15 to 20; mean divergence value 17.06) (Table 5). No relation was observed between CFSV and any other Pestivirus species (Fig. 2). Table 1. List of Pestivirus strains of CSFV species (n=126) evaluated according to palindromic secondary structure characteristics at the RNA 5′-UTR (PNS method). The nomenclature of identified genotypes is based on divergence in the genus. Clustering according to the primary sequence analysis by depositors is indicated under parenthesis. ND: not determined. Cattle (B. taurus); Pig (Sus scrofa domesticus); Sheep (Ovis aries); Wild boar (Sus scrofa scrofa).
Table 2. Alignment of genus Pestivirus CSFV species genotypes variable loci 5′-UTR RNA secondary structure sequences, segregated according to types of base pair combinations. Base pairing combinations were identified for genotype characterization (n=37) and considered for identification marker definition. The different types are ordered according to increasing divergence in the species (*), and divergence in the genus (**) was expressed in a number of divergent base pairs, with reference to most common base pairs in the prevalent positions. Highly conserved base pair positions are excluded. Positions 13 in V1 locus and 2, 7 and 8 in V3 locus are species specific; positions 15 in V1 locus, 5 and 7 in V2 locus and 1 in V3 locus are genotype specific. Y: C or U; ND: not determined.
By phylogenetic analysis of E2 region, the Indian bovine strains CS/ML/911/IDP/13 (KY860532) and CS/ML/AF/Umiam/14 (KY860531) (Ahuja et al., 2015; Chakraborty et al., 2018) were similar to pig isolates as IND/AS/GHY/G4 (KM362426) (Ahuja et al., 2015), originated from Assam, a neighboring state. By BLAST, the E2 sequence of both strains CS/ML/911/IDP/13 and CS/ML/AF/Umiam/14 showed 99% and 92% nucleotide identity with strain IND/AS/GHY/G4 (CSFV-a5) and reference strain Paderborn (GQ902941) (CSFV-a2), respectively. Taking into account the PNS evaluation of Paderborn and IND/AS/GHY/G4 5′-UTR, both bovine strains CS/ML/911/IDP/13 and CS/ML/AF/Umiam/14 were clustered into CSFV-a5. Similar to type Parambi (CSFV-a4), also this cluster was the characteristic of India. A schematic phylogenetic tree, based on secondary structure base pair variation and divergence values at the three variable loci, is shown in Figure 3. At the species level, the observed taxonomic status of the examined strains corresponded to the estimation obtained by phylogenetic tree based on the primary sequence analysis of the 5′-UTR constructed from the alignment with the representative strains from the identified genogroups (Fig. 4). DiscussionOn contrary to other pestiviruses, as BVDV-1 which accounted for much more genotypic variants (Giangaspero and Apicella, 2018), the CSFV species was low heterogeneous. Only two genotypes, CSFV-c and CSFV-d, both of Asian origin, were highly divergent. Taking into account the high divergence in the species based on the primary sequence analysis, the strain Kanagawa/74 (CSFV-c) (Japan, 1974; Dreier et al., 2007; Beer et al., 2015) was proposed, similarly to congenital Tremor (United Kingdom, 1964), as harmonized outgroup strain for phylogenetic analyses (Paton et al., 2000). Similar to the strain Kanagawa/74, the Chinese bovine strain S171 (CSFV-d) was divergent in the species (Fig. 4), suggesting its consideration as another outgroup of the species with even more enhanced divergence. The other bovine Chinese strains are less divergent, showing common genomic trait characteristic of the known reported CSFV-a2 strains, indicating the concomitant circulation of typical CSV types with others showing atypical characteristics, possibly related to the host species adaptation or a result of geographic isolation. Such occurrences have been suggested also for BVDV-1 isolates in China (Xue et al., 2010; Wang et al., 2014). Similarly, Indian bovine strains were genetically closely related to common CSFV genotype a1 strains and corresponded to circulate Pestivirus pig strains reported in the country. Between 2009 and 2011, Rajkhowa et al. reported three CSFV isolates from pigs (Aizawl-09, 5NCR/CSF/MZ/AIZ/348, and 5NCR/CSF/MZ/AIZ/352) (accessions numbers HM449066, JX975460, and JX975461, respectively), from the Indian state of Mizoram, located at the extreme northeastern border of Myanmar, well distant from Tamil Nadu, the area where CSFV was detected in cattle. Pig strains from Mizoram are very close to the bovine strains, belonging to the genotype CSFV-a variant 1. A distinctive guanine uracile base pairing in position 3 in the V1 locus was found only in the bovine isolates originated from Tamil Nadu. This peculiarity was different from all other CSFV strains, which present a conserved A–U pairing in this position. Interestingly, G-C or G*U pairings are conserved only among strains belonging to the BVDV species types 1, 2, and 3 (only five exceptions, out of more than 1,000 considered sequences). In the CSFV species, three genetic clusters referred specifically to Asian countries. The CSFV genotype a variant 4 (type Parambi) included only pig and wild boar strains from India [Bhaskar et al., 2015; and sequences deposited by Ravishankar et al. (unpublished); Tomar et al. (unpublished); and Chandramohan et al. (unpublished)]. CSFV genotype C (type Okinawa) was reported only in pigs originating from Japan and Taiwan (Harasawa and Giangaspero, 1999; Sakoda et al., 1999; Lin et al., 2007; and strain P97 deposited by Liu). Furthermore, the sequence characteristics of strain S171 (Zhang et al., 2014) (CSFV-d) were observed only in China. Table 3. Clustering into genotypes of Pestivirus CSFV species strains (n=126) evaluated according to the PNS method at the 5’ untranslated region of RNA. Nomenclature according to primary sequence analysis is indicated under parenthesis. ND: not determined.
Table 4. PNS characteristics to the Pestivirus CSFV species with related genotypes and genotype variants. The position of base pairings is defined by numbering from the bottom of the variable locus. Y=C or U.
In general, apart nomenclature difference, the comparison between primary and secondary structure analyses revealed corresponding main genomic groups, defined by phylogenetic analysis, dividing CSFVs into three major lineages (Brescia, Alfort and Kanagawa/Okinawa types) and their sublineages (Lin et al., 2007). Furthermore, the phylogenetic trees showed such similarity (Figs. 3 and 4). Genotype CSFV-a variant 1 (type Brescia) corresponded to 1.1 and 1.2, CSFV-a variant 2 (type Alfort/Tübingen) to 2.1 and 2.3, CSFV-a variant 4 to 2.2, and genotype CSFV-c to 3 and 3.4. The CSFV 2.2 strain Sch180 (JQ411560) from Germany and the CSFV 1.3 strain VRI 4167 (JQ411570) from Malaysia could not be determined due to incomplete V1 locus, but their other sequence fragments were compatible with PNS genotype variants CSFV-a3 (type Saitama) and CSFV-a5 (type Honduras), respectively (Table 2). However, as expected, the specific differences observable at the level of the E2 gene sequence fragment (Risatti et al., 2005; Postel et al., 2012) were not necessarily related to secondary structure 5′-UTR clustering. Similarly, PNS genotype CSFV-c included variants as 3.4 not discriminable from other members of genotype 3 (Lin et al., 2007). Similar discrepancies may occur comparing other regions, as between 5′-UTR and Npro.
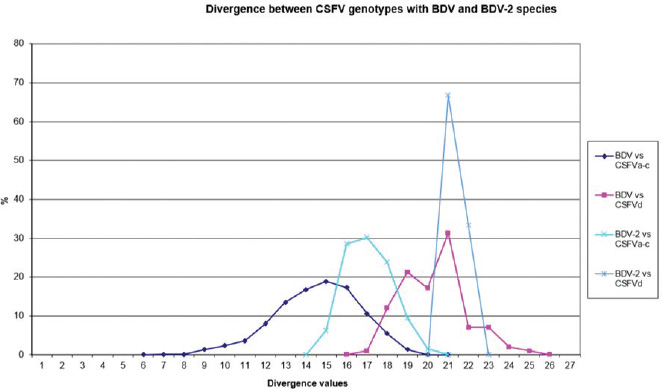
Fig. 1. Graphical representation of PNS quantitative evaluation. CSFV shows close genetic relation to BDV and a lesser extent with BDV-2. With concern to the relationship between CSFV detection and clinical observations, the strain HEN03, the only among the Chinese strains for which clinical anamnestic data could be retrieved, was associated with reproductive disorders observed in many animals in the herd: abortion, stillbirth, and reduced milk yield. Furthermore, the mortality rate was high. However, concomitant infection with BVDV-1 and BVDV-2 was revealed. It is particularly interesting that also all the Indian bovine CSFV strains were isolated in herds suffering from reproductive disturbances, abortions, birth defects, repeat breeding, and mastitis. Serological positivity for bovine herpesvirus 1 (IBR) and Brucella sp. was absent or very low, manifestly not correlated to observed pathologies, and no BVDV has been isolated (Giangaspero et al., 2017). Hypothetically, CSFV bovine strains detected in China and India might find their origin from vaccine prophylaxis performed in pig populations. CSFV-a1 comprises a CSFV-lapinized vaccine strain from India (EU857642–AF091507), which showed a high homology with Indian cattle strains (99% identities according to the results obtained by BLAST). Investigations on vaccination protocols applied in the research area showed that none of the cattle farms investigated in Tamil Nadu showed to have contact with pigs, and no immunization program was carried for BVDV (Giangaspero et al., 2017). However, CSFV vaccine prophylaxis is carried out in the swine industry in India. At present, anti-BVDV-killed virus vaccines are available in China, but, for a long time, there were no commercial BVDV vaccines in the Chinese market. In China, and particularly in Tibet and Qinghai provinces, prophylaxis against BVDV in cattle and yaks was based on the wide use of the live-attenuated hog cholera lapinized vaccine (HCLV), administered to animals at a triple dose. This followed the experimental demonstration of efficacy and safety of the prophylactic use of HCLV for BVDV in lactating and pregnant cows, calves, and yaks undertaken by Yuan Qingzhi in 1957 (Liu et al., 2003). For a long time, the HCLV was the only authorized vaccine against CSFV in China. In Tibet, the long-term use of live-attenuated HCLV not only reduced losses caused by BVDV infection but also gave the opportunity for vaccinal strain adaptation in cattle, allowing as a possible consequence of a subsequent natural diffusion. Tibet is historically a high-quality breeding region managed by a traditional seminomadic agropastoral system and appreciated by traders who seasonally purchase and move live animals and may be possibly responsible for the spread of pestiviruses, especially through immunotolerant persistently infected subjects. In Spain, the reported ovine strain 5440/99 showed the sequence similarities with CSFV vaccine strains used in pigs, suggesting spillovers from prophylactic campaigns (Hurtado et al., 2003). In Belgium, some fetal calf sera positive for CSFV-like 5′-UTR sequences, experimentally not infecting or seroconverting pigs, have been detected during the 90s (Dr Koenen, Sciensano, Belgium, personal communication), suggesting circulation of unidentified virus strains with sequence similarity to CSFV within the 5′-UTR. However, in Belgium, since the last occurrence of CSFV was reported in 1997, thus, in this period, wild and vaccinal strains were present in the country, and it cannot be excluded from the occurrence of possible spillovers in cattle from CSF-infected or immunized pigs. Table 5. PNS quantitative evaluation. CSFV versus BDV and BDV-2 tentative species secondary structure sequence comparison divergence values (n=2,178). Scores exceeding species determination divergence limit value (13) indicate genetic distance and different species clustering.
Fig. 2. Graphical representation of PNS quantitative evaluation. Genotype CSFV-d is not related to BDV or BDV-2.
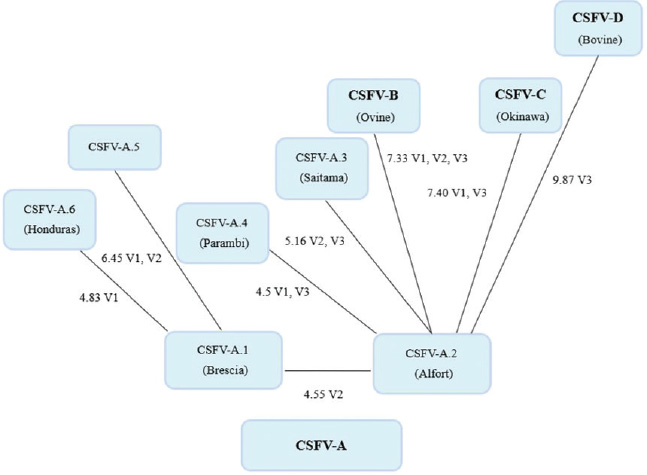
Fig. 3. Schematic phylogenetic tree based on nucleotide changes in the secondary structure of classical swine fever virus genotypes. Mean values of base–pair divergence and variable locus location indicate the evolutionary changes.
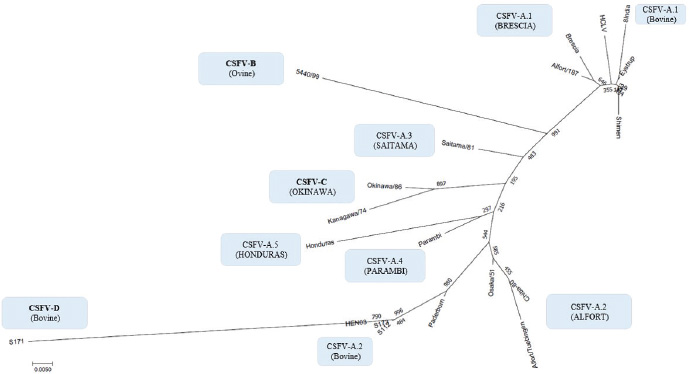
Fig. 4. Phylogenetic tree based on the 5′-UTR comparison, showing taxonomic relationships of the CSFV strains in the Pestivirus genus. Strains Brescia (M31768), 39 (AF407339), Eystrup (AF326963), Alfort/187 (X87939), HCLV (AF091507), Shimen (AF092448), and the bovine strain 8 India (MG813566) are references for the genotype CSFV-a variant 1. Strains Alfort/Tuebingen (J04358), Chiba-80 (AB019659), Osaka/51 (AB019174), Paderborn (AY072924), and the Chinese bovine strains S173 (KF006975), HEN03 (KC176778), and S112 (MK118725) are references for the genotype CSFV-a variant 2. Strain Saitama/81 (AB019178) is the reference for the CSFV-a variant 3, and strains Parambi (KT239105), IND/AS/GHY/G4 (KM362426), and Honduras (L42426) are the references for the CSFV-a variants 4, 5, and 6, respectively. The Spanish ovine strain 5440/99 (AY159514) is the reference for the genotype CSFV-b. The Japanese strains Kanagawa/74 (AB019166) and Okinawa/86 (AB019172) are references for the genotype CSFV-c. The Chinese bovine strain S171 (KF006974) represents the genotype CSFV-d. Distances were computed using Clustal X, version 1.8, using the neighbor-joining method. Scale bar indicates 10 nucleotide substitutions per 100 nucleotides. Nomenclature of identified genotypes is based on divergence in the genus. The recent taxonomy of CSFV is based on the analysis of the E2 genomic region (Postel et al., 2012). Due to the relation to envelope glycoprotein (the main immunogen of CSFV, thus essential for the development of vaccines) (Perez et al., 2012) and the longer size (1,119 nucleotides), the full-length E2 encoding sequences were recommended for accurate phylogenetic analyses, assessing the viral types and subtypes. Based on the primary sequence analysis, three CSFV genotypes, each with three or six subgroups, have been identified (Paton et al., 2000; Dreier et al., 2007; Postel et al., 2013; Beer et al., 2015; Silva et al., 2017). However, despite a limiting factor for the differentiation of closely related isolates (Postel et al., 2012), short-length target fragments in other regions of the viral genome as 5′-UTR or NS5B have been largely applied for genotyping purposes and molecular epidemiology (Lowings et al., 1996; Paton et al., 2000; Beer et al., 2015). Phylogenetic analysis of the sequences based on a 190-nucleotide (nt) fragment of the E2 glycoprotein proved very useful for the genetic typing of isolates in the case of new outbreaks (Dreier et al., 2007). In any case, laboratory testing international standards are indicated only by the World Organization for Animal Health (Office International des Epizooties - OIE). According to the Terrestrial Manual of the World Organization for Animal Health (Office International des Epizooties—OIE) (Chapter 2.8.3 CSFV; Paragraph 1.1.5.) (OIE, 2014), as the E2 major glycoprotein gene (190 nucleotides), the fragment 5’-UTR of the CSFV genome (150 nucleotides), targeted for molecular characterization of the bovine sequences in the present study, is admitted too for genetic typing and molecular epidemiology of CSF, based on the comparison of genetic differences between virus isolates. According to the Terrestrial Code (Chapter 15.2; Article 15.2.28, Paragraph 3, Virological surveillance) (OIE, 2017) and the Terrestrial Manual (Chapter 2.8.3; Paragraph 1.1.4.) (OIE, 2014) of the World Organization for Animal Health, the RT-PCR amplification of CSFV RNA followed by nucleotide sequencing, applied for the obtention of the sequences evaluated in the present study (Zhang et al., 2014; Giangaspero et al., 2017), originally focused on investigations of BVDV, is admitted among recommended methods for CSFV agent detection, confirmation of clinical cases, and differentiation from other pestiviruses. It is clear that the sequence of the gene encoding the E2 glycoprotein is preferred in the recent taxonomy of the species (Postel et al., 2012), but the consideration of 5′-UTR was the only possible approach to perform a comparative evaluation of CSFV with sequences of the other different species, otherwise not feasible taking into account that the majority of Pestivirus-deposited sequences are 5′-UTR (Yesilbag et al., 2017; Giangaspero et al., 2018). The secondary structure–sequence characterization allowed an exhaustive comparative evaluation with genomic variants in the genus, about 1,400 Pestivirus strain sequences. The particularity of the PNS method is the exclusive consideration of strategic genomic sequences corresponding to the 5’-UTR IRES, which is responsible for translational, transcriptional, and replicational events in pestiviruses. Thus, at this level, stable nucleotide variations assume a high importance in terms of virus evolutionary history. The PNS method has been successfully applied for the characterization of pestiviruses, including atypical species as Giraffe, Pronghorn, or Bungowannah (Harasawa et al., 2000; Giangaspero and Harasawa, 2008; 2011), thus adequate for the scope of the present study. The presence of CSFV 5′-UTR sequences in bovines from India and China appeared atypical. However, another recent report from India (Chakraborty et al., 2018) strongly corroborated the previous observations, amplifying also full-length E2 region of CSFV in two positive samples, and suggesting a potential emerging health risk in cattle. Furthermore, the possible occurrence of similar circulation in cattle in CSFV endemic environment in Kenya is currently under evaluation (Prof VanLeeuwen, University of Prince Edward Island, Canada, personal communication). ConclusionsThese preliminary observations are new and relevant in countries, where CSFV control and eradication strategies are applied. Further investigations will be necessary to confirm adaptation and diffusion of the virus in cattle as well as provide a clear picture of genetic characteristics, in particular, to evaluate other genomic regions, E2, and eventually full-length genome, and apply other internationally recognized tests. Furthermore, it will be important to clarify the clinical suspicions and related virus differential diagnosis for correct notification obligations in compliance with the World Organization for Animal Health reference norms. Conflict of interestAuthors declare that there is no conflict of interest. Authors’ contributionM. Giangaspero and S. Zhang contributed equally to the present study. ReferencesAhuja, A., Bhattacharjee, U., Chakraborty, A.K., Karam, A., Ghatak, S., Puro, K., Das, S., Shakuntala, I., Srivastava, N., Ngachan, S.V. and Sen A. 2015. Complete genome sequence of classical Swine Fever virus subgenogroup 2.1 from assam, India. Genome Announc. 3 (1). Bhaskar, N., Ravishankar, C., Rajasekhar, R., Sumod, K., Sumithra, T.G,, John, K., Mini, M., Ravindran, R., Shaji, S. and Aishwarya, J. 2015. Molecular typing and phylogenetic analysis of classical swine fever virus isolates from Kerala, India. Virus Dis. 26 (4), 260–266. Beer, M., Goller, K.V., Staubach, C. and Blome, S. 2015. Genetic variability and distribution of Classical swine fever virus. Anim. Health Res. Rev. 16 (1), 33–39. Biró, J., Oláh, P. and Palatka, Z. 1966. Attempt to adapt the lapinized hog cholera virus strain “Suvac” to sheep. Acta Vet. Acad. Sci. Hung. 16(3), 293–299. Center for Food Security and Public Health (CFSPH). 2015. Classical Swine Fever. Factsheets. Available via www.cfsph.iastate.edu Chakraborty, A.K., Karam, A., Mukherjee, P., Barkalita, L., Borah, P., Das, S., Sanjukta, R., Puro, K., Ghatak, S., Shakuntala, I., Sharma, I., Laha, R.G. and Sen, A. 2018. Detection of classical swine fever virus E2 gene in cattle serum samples from cattle herds of Meghalaya. Virus Dis. 29(1), 89–95. Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T.J., Higgins, D.G. and Thompson, J.D. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500. Dreier, S., Zimmermann, B., Moennig, V. and Wilke, I.G., 2007. A sequencedatabase allowing automated genotyping of classical swine fevervirus isolates. J. Virol. Methods 140, 95–99. Giangaspero, M. and Apicella, C. 2014. Improved Palindromic Nucleotide Substitutions software version 2.0. Genotyping based on the secondary structure alignment in the 5’ untranslated region of Pestivirus RNA. J. Bioinfor. Intel. Cont. 3(1), 39–64. Giangaspero, M. and Apicella, C. 2018. Bovine viral diarrhea virus type 1 current taxonomy according to palindromic nucleotide substitutions method. J. Virol. Methods 256, 37–76. Giangaspero, M. and Harasawa, R. 2007. Numerical taxonomy of genus Pestivirus based on palindromic nucleotide substitutions in the 5’ untranslated region. J. Virol. Methods 146, 375–388. Giangaspero, M. and Harasawa, R. 2008. Genetic variation of classical swine fever virus based on palindromic nucleotide substitutions, a genetic marker in the 5’ untranslated region of RNA. Vet. Ital. 44(2), 305–318. Giangaspero, M. and Harasawa, R. 2011. Species Characterization in the genus Pestivirus according to Palindromic Nucleotide Substitutions in the 5’ Untranslated Region. J. Virol. Methods 174(1–2), 166–172. Giangaspero, M., Kumar, S.K. and Zhang, S.Q. 2017. Classical swine fever virus in cattle. Vet. Rec. 181(3), 73. Giangaspero, M., Yesilbag, K. and Apicella, C. 2018. Who’s who in the Bovine viral diarrhea virus type 1 species: genotypes L and R. Virus Res. 256, 50–75. Gong, W., Zhang, L., Lu, Z., Jia, J., Wang, M., Peng, Z., Guo, H., Shi, J. and Tu, C. 2016. Complete genome sequence of a novel sub-subgenotype 2.1g isolate of classical swine fever virus from China. Arch. Virol. 161(9), 2613–2617. Harasawa, R. and Giangaspero, M. 1998. A novel method for pestivirus genotyping based on palindromic nucleotide substitutions in the 5’-untranslated region. J. Virol. Methods 70, 225–230. Harasawa, R. and Giangaspero, M. 1999. Genetic variation in the 5’end and NS5B regions of classical swine fever virus genome among Japanese isolates. Microbiol. Immunol. 43(4), 373–379. Harasawa, R., Giangaspero, M., Ibata, G. and Paton, P.J. 2000. Giraffe strain of pestivirus. Its taxonomic status based on the 5’ untranslated region. Microbiol. Immunol. 44(11), 915–921. Hurtado, A., Garcia-Perez, A.L., Aduriz, G. and Juste, R.A. 2003. Genetic diversity of ruminant pestiviruses from Spain. Virus Res. 92, 67–73. Jiang, D.L., Liu, G.H., Gong, W.J., Li, R.C., Hu, Y.F., Tu, C. and Yu, X.L. 2013. Complete genome sequences of classical Swine Fever virus isolates belonging to a new subgenotype, 2.1c, from hunan province, china. Genome Announc. 1(1), E000 80–12. Kamboj, A., Patel, C.L., Chaturvedi, V.K., Saini, M. and Gupta, P.K. 2014. Complete genome sequence of an Indian field isolate of classical Swine Fever virus belonging to subgenotype 1.1. Genome Announc. 2(5). Kumar, S., Stecher, G. and Tamura, K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. Leifer, I., Hoffmann, B., Hoper, D., Bruun Rasmussen, T., Blome, S., Strebelow, G., Horeth-Bontgen, D., Staubach, C. and Beer, M. 2010. Molecular epidemiology of current classical swine fever virus isolates of wild boar in Germany. J. Gen. Virol. 91(11), 2687–2697. Li, X., Xu, Z., He, Y., Yao, Q., Zhang, K., Jin, M., Chen, H. and Qian, P. 2006. Genome comparison of a novel classical swine fever virus isolated in China in 2004 with other CSFV strains. Virus Genes 33(2), 133–142. Lin, Y.J., Chien, M.S., Deng, M.C. and Huang, C.C. 2007. Complete sequence of a subgroup 3.4 strain of classical swine fever virus from Taiwan. Virus Genes 35(3), 737–744. Liu, Y., Yin, Z., Liu, S., Han, P., Hua, S., Yu, Y., Shi, Q., Ding, X., Liu, X. and Yu, R. 2003. Study on the prevention of bovine viral diarrhoea-mucosal disease of the yak. Chinese J. Prev. Vet. Med. 6. Loan, R.W. and Storm, M.M. 1968. Propagation and transmission of hog cholera virus in non-porcine hosts. Am. J. Vet. Res. 29, 807–811. Lowings, P., Ibata, G., Needham, J. and Paton, D. 1996. Classical swine fever virus diversity and evolution. J. Gen. Virol. 77, 1311–1321. Mohamed, T.M. 2004. BVD Report UAE, 2004. Available via https://www.researchgate.net/publication/236141896_BVD_ReportUAE2004 (Accessed 8 August 2019). Patil, S.S., Hemadri, D., Shankar, B.P., Raghavendra, A.G., Veeresh, H., Sindhoora, B., Chandan, S., Sreekala, K., Gajendragad, M.R. and Prabhudas, K. 2010. Genetic typing of recent classical swine fever isolates from India. Curr. Microbiol. 141(3–4), 367–373. Paton, D.J., McGoldrick, A., Greiser-Wilke, I., Parchariyanon, S., Song, J.Y., Liou, P.P., Stadejek, T., Lowings, J.P., Björklund, H. and Belák, S. 2000. Genetic typing of classical swine fever virus. Vet. Microbiol. 73(2–3), 137–157. Perez, L.J., Arce, H.D., Perera, C.L., Rosell, R., Frias, M.T., Percedo, M.I., Tarradas, J., Dominguez, P., Nunez, J.I. and Ganges, L. 2012. Positive selection pressure on the B/C domains of the E2-gene of classical swine fever virus in endemic areas under C-strain vaccination. Infect. Genet. Evol. 12, 1405–1412. Postel, A., Schmeiser, S., Bernau, J., Meindl-Boehmer, A., Pridotkas, G., Dirbakova, Z., Mojzis, M. and Becher, P. 2012. Improved strategy for phylogenetic analysis of classical swine fever virus based on full-length E2 encoding sequences. Vet. Res. 43(1), 50. Postel, A., Schmeiser, S., Perera, C.L., Rodrıguez, L.J., Frias-Lepoureau, M.T. and Becher, P. 2013. Classical swine fever virus isolates from Cuba form a new subgenotype 1.4. Vet. Microbiol. 161, 334–338. Risatti, G.R., Borca, M.V., Kutish, G.F., Lu, Z., Holinka, L.G., French, R.A., Tulman, E.R. and Rock, D.L. 2005. The E2 glycoprotein of classical swine fever virus is a virulence determinant in swine. J. Virol. 79(6), 3787–3796. Saitou, N. and Nei, M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. Sakoda, Y., Ozawa, S., Damrongwatanapokin, S., Sato, M., Ishikawa, K. and Fukusho, A. 1999. Genetic heterogeneity of porcine and ruminant pestiviruses mainly isolated in Japan. Vet. Microbiol. 65(1), 75–86. Shen, H., Pei, J., Bai, J., Zhao, M., Ju, C., Yi, L., Kang, Y., Zhang, X., Chen, L., Li, Y., Wang, J. and Chen, J. 2011. Genetic diversity and positive selection analysis of classical swine fever virus isolates in south China. Virus Genes 43(2), 234–242. Shimizu, M. and Kumagai, T. 1989. Experimental infection of pregnant goats with swine fever virus. Vet. Microbiol. 20, 207–214. Silva, M.N., Silva, D.M.F., Leite, A.S., Gomes, A.L.V., Freitas, A.C., Pinheiro-Junior, J.W., Castro, R.S. and Jesus, A.L.S. 2017. Identification and genetic characterization of classical swine fever virus isolates in Brazil: a new subgenotype. Arch. Virol. 162(3), 817–822. Tomar, N., Gupta, A., Arya, R.S., Somvanshi, R., Sharma, V. and Saikumar, G. 2015. Genome sequence of classical Swine Fever virus genotype 1.1 with a genetic marker of attenuation detected in a continuous porcine cell line. Genome Announc. 3(2). Wang, W., Shi, X., Tong, Q., Wu, Y., Xia, M.Q., Ji, Y., Xue, W. and Wu, H. 2014. A bovine viral diarrhea virus type 1a strain in China: isolation, identification, and experimental infection in calves. Virol. J. 11, 8. World Organisation for Animal Health (OIE). 2014. Terrestrial Manual Chapter 2.8.3. World Organisation for Animal Health (OIE). 2017. Terrestrial Code, Chapter 15.2. Xue, F., Zhu, Y.M., Li, J., Zhu, L.C., Ren, X.G., Feng, J.K., Shi, H.F. and Gao, Y.R. 2010. Genotyping of bovine viral diarrhea viruses from cattle in China between 2005 and 2008. Vet. Microbiol. 143(2–4), 379–383. Yesilbag, K., Alpay, G. and Becher, P. 2017. Variability and global distribution of subgenotypes of bovine viral diarrhea virus. Viruses 9(6), 128. Zhang, S.Q., Tan, B., Guo, L., Wang, F.X., Zhu, H.W., Wen, Y.J. and Cheng, S. 2014. Genetic diversity of bovine viral diarrhea viruses in commercial bovine serum batches of Chinese origin. Infect. Genet. Evol. 27, 230–233. Zhou, W., Gao, S., Podgorska, K., Stadejek, T., Qiu, H.J., Yin, H., Drew, T. and Liu, L. 2014. Rovac is the possible ancestor of the Russian lapinized vaccines LK-VNIVViM and CS strains but not the Chinese strain (C-strain) vaccine against classical swine fever. Vaccine 32(49), 6639–6642. | ||
| How to Cite this Article |
| Pubmed Style Giangaspero M, Zhang SQ. Genomic Characteristics of Classical Swine Fever Virus Strains of Bovine Origin According to Primary and Secondary Sequence Structure Analysis. Open Vet J. 2020; 10(1): 94-115. doi:10.4314/ovj.v10i1.15 Web Style Giangaspero M, Zhang SQ. Genomic Characteristics of Classical Swine Fever Virus Strains of Bovine Origin According to Primary and Secondary Sequence Structure Analysis. https://www.openveterinaryjournal.com/?mno=79845 [Access: July 10, 2025]. doi:10.4314/ovj.v10i1.15 AMA (American Medical Association) Style Giangaspero M, Zhang SQ. Genomic Characteristics of Classical Swine Fever Virus Strains of Bovine Origin According to Primary and Secondary Sequence Structure Analysis. Open Vet J. 2020; 10(1): 94-115. doi:10.4314/ovj.v10i1.15 Vancouver/ICMJE Style Giangaspero M, Zhang SQ. Genomic Characteristics of Classical Swine Fever Virus Strains of Bovine Origin According to Primary and Secondary Sequence Structure Analysis. Open Vet J. (2020), [cited July 10, 2025]; 10(1): 94-115. doi:10.4314/ovj.v10i1.15 Harvard Style Giangaspero, M. & Zhang, . S. Q. (2020) Genomic Characteristics of Classical Swine Fever Virus Strains of Bovine Origin According to Primary and Secondary Sequence Structure Analysis. Open Vet J, 10 (1), 94-115. doi:10.4314/ovj.v10i1.15 Turabian Style Giangaspero, Massimo, and Shu Qin Zhang. 2020. Genomic Characteristics of Classical Swine Fever Virus Strains of Bovine Origin According to Primary and Secondary Sequence Structure Analysis. Open Veterinary Journal, 10 (1), 94-115. doi:10.4314/ovj.v10i1.15 Chicago Style Giangaspero, Massimo, and Shu Qin Zhang. "Genomic Characteristics of Classical Swine Fever Virus Strains of Bovine Origin According to Primary and Secondary Sequence Structure Analysis." Open Veterinary Journal 10 (2020), 94-115. doi:10.4314/ovj.v10i1.15 MLA (The Modern Language Association) Style Giangaspero, Massimo, and Shu Qin Zhang. "Genomic Characteristics of Classical Swine Fever Virus Strains of Bovine Origin According to Primary and Secondary Sequence Structure Analysis." Open Veterinary Journal 10.1 (2020), 94-115. Print. doi:10.4314/ovj.v10i1.15 APA (American Psychological Association) Style Giangaspero, M. & Zhang, . S. Q. (2020) Genomic Characteristics of Classical Swine Fever Virus Strains of Bovine Origin According to Primary and Secondary Sequence Structure Analysis. Open Veterinary Journal, 10 (1), 94-115. doi:10.4314/ovj.v10i1.15 |

