




| Research Article | ||
Open Vet J. 2023; 13(7): 903-931 Open Veterinary Journal, (2023), Vol. 13(7): 903-931 Original Research Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and TurkeyMassimo Giangaspero1* and Shuquin Zhang21Faculty of Veterinary Medicine, University of Teramo, Teramo, Italy 2Institute of Special Animal and Plant Sciences, Chinese Academy of Agricultural Sciences, Changchun, People’s Republic of China *Corresponding Author: Massimo Giangaspero. Faculty of Veterinary Medicine, University of Teramo, Teramo, Italy. Email: giangasp [at] gmail.com Submitted: 14/11/2022 Accepted: 27/06/2023 Published: 31/07/2023 © 2023 Open Veterinary Journal
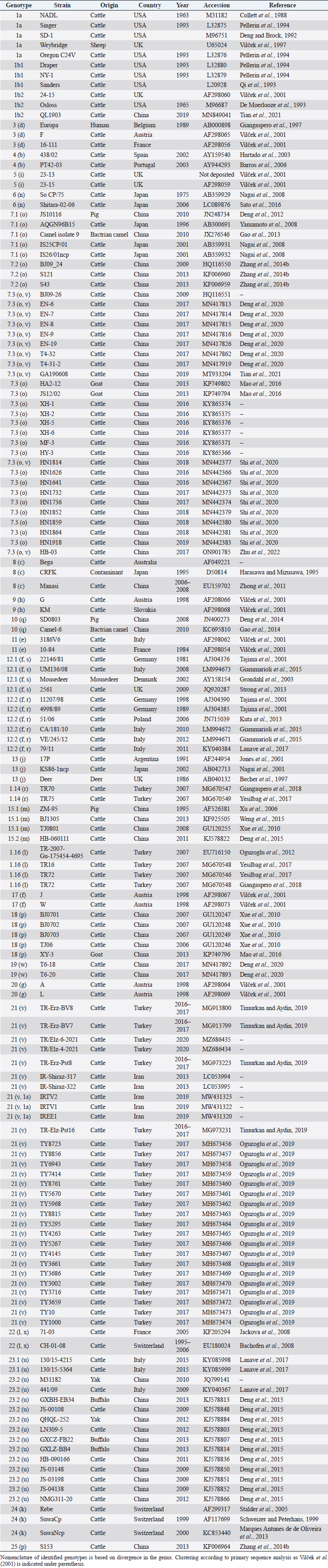
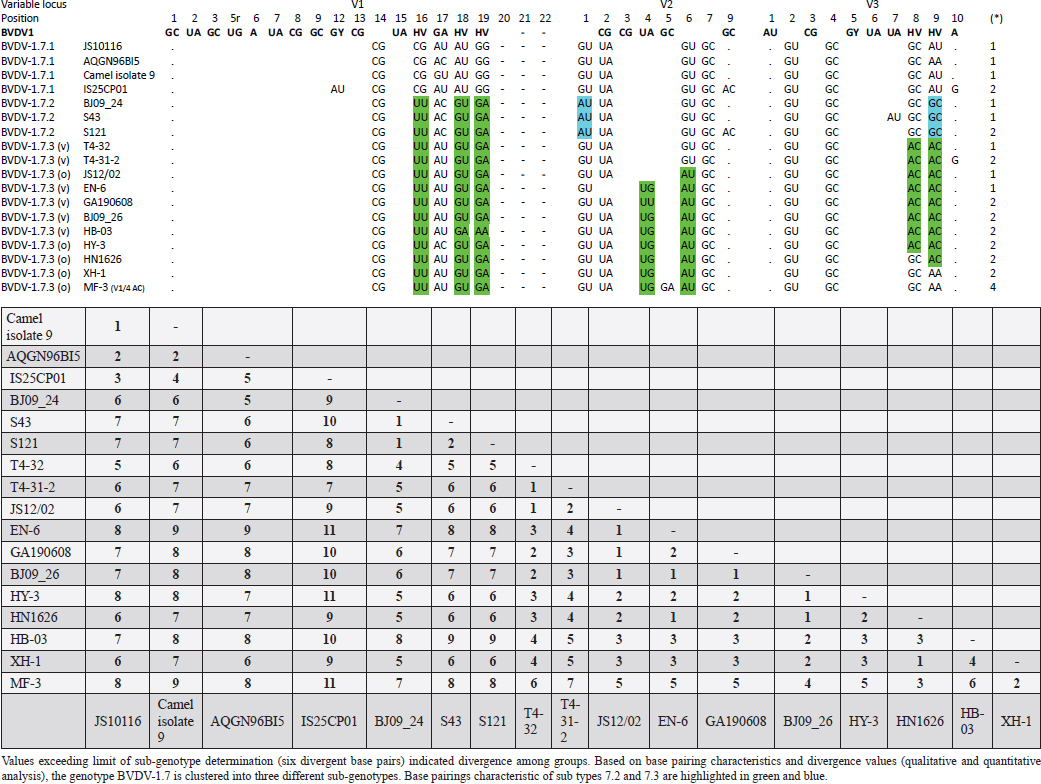
AbstractBackground: Pestivirus A Bovine viral diarrhea virus type 1 (BVDV-1) is a heterogeneous species within the genus, affecting cattle and other ruminants, with economic impact on livestock production. Aim: The study aimed to update the taxonomy of the Pestivirus A, BVDV-1 species and to verify the clustering of the strains reported as genotype 1v, originating from different countries. Methods: Recently deposited strains from China, Turkey, and Iran have been evaluated by the palindromic nucleotide substitutions (PNS) genotyping method. Results: Based on secondary structure analysis of the 5′-UTR sequences, strains reported as 1v from China were clustered as sub genotype 1.7.3 (1o). Genotype 1.19 (1w) was restricted to China and genotype 1.21 (1v) was present only in Turkey and Iran. Conclusion: The application of the PNS method clarified the taxonomical status of strains, revealing the homonymy of genetically different clusters. Furthermore, these observations indicated geographic segregation in the Pestivirus A species, and confirmed the occurrence of new atypical genetic variants, with potential implications on control and prophylaxis. Keywords: Asia, Bovine viral diarrhea virus type 1, Geographic segregation, Pestivirus, Taxonomy. IntroductionPestivirus A Bovine viral diarrhea virus type 1 (BVDV-1) is an established species of the genus Pestivirus of the family Flaviviridae, responsible for a disease with widespread distribution, affecting cattle and other ruminants, presenting a wide range of clinical manifestations, with a substantial economic impact on livestock production. Currently, it is difficult to obtain an exhaustive comparison of all variants in the BVDV-1 species by full-length genome analysis, giving that about 97% of deposited sequences are restricted to genome short portions (5′-UTR mainly) (Yesilbag et al., 2017). Therefore, genotypes are characterized predominantly by the analysis of the 5′-UTR. While the E2 gene sequence is considered relevant for accurate typing of classical swine fever virus (CSFV) (Postel et al., 2012), this region is rarely applied to BVDV-1 strains and does not provide additional information in terms of taxonomy. Also, Npro genomic region is used to characterize BVDV isolates and results generally correspond to the clustering based on 5′-UTR and no additional variants in the species were detected with other analytic techniques evaluating other genomic regions (Giangaspero et al., 2018). BVDV is categorized genetically on the base of nucleotide sequence variations. Strains (field isolates) have been segregated into genotypes by different authors. Initially, genotypes BVDV-1a and BVDV-1b have been reported (Harasawa, 1996). Furthermore, genotypes BVDV-1c and BVDV-1d have been described (Giangaspero et al., 1997, 2001). The evaluation of other isolates, by sequence alignment and construction of phylogenetic trees, demonstrated the presence of other genotypes within the BVDV-1 species (Baule et al., 1997; Tajima et al., 2001; Vilček et al., 2001; Couvreur et al., 2002; Ciulli et al., 2003; Hurtado et al., 2003). Also sub genotypes have been defined, for example, genotype BVDV-1a was subdivided into a1 and a2 (Bachofen et al., 2008) or BVDV-1b into b1 and b2 (Tajima et al., 2001). According to secondary structure, limit values of a number of divergent base pairs (bps) have been identified to determine genotypes or sub-genotypes (Giangaspero and Harasawa, 2007). Another characteristic of the field isolates is their capacity to induce or not cytopathic effect on cellular substrate (disruption of cells) when grown in tissue culture for laboratory investigations. Thus, strains are distinguished into two biotypes: non-cytopathic and cytopathic. The non-cytopathic strains are predominant in nature and important for clinical disease (Rajput et al., 2017). In recent years, a relatively large number of new sequences of isolates, mainly from domestic animals, have been deposited in genetic sequence databases, in particular from Asian countries, and additional BVDV-1 genotypes were reported describing atypical variants within the species (Xue et al., 2010; Zhong et al., 2011; Gao et al., 2013; Wang et al., 2014; Zhang et al., 2014b; Deng et al., 2015; Weng et al., 2015; Zhu et al., 2016). A certain confusion in the nomenclature of the increasing number of genotypes was evident and it is still a matter of concern, indicating the need for harmonization. Homonymy of BVDV-1 genotypes, as 1l or 1r applied to strains from different genetic clusters, was previously reported (Yesilbag et al., 2017; Giangaspero et al., 2018). More recently, strains collected from cattle in China in 2017 were reported as belonging to two unknown groups, tentatively typed as “BVDV-1v” and “BVDV-1w” (Deng et al., 2020). Deng clustered as 1v also the two goat strains JS12/02 and HA2-12 from China (Deng et al., 2020). However, these stains have been previously reported as 1o (Mao et al., 2016). In 2019, a non-cytopathic strain of BVDV was isolated from cattle and named GA190608 (Tian et al., 2021). Phylogenetic analysis based on the 5′-UTR sequence revealed that the BVDV isolate GA190608 clustered with strains HN1814, EN-19, and BJ09_26 in a separate branch, which has tentatively been classified as a new genetic subtype, "1v." The strain EN-19 was reported as 1v (Deng et al., 2020), the strain HN1814 was reported as 1o (Shi et al., 2020) and the strain BJ09_26 was deposited without a defined genotype. Furthermore, during 2016 and 2020, virus strain sequences were deposited as 1l or 1v from cattle isolates from Turkey (Oguzoglu et al., 2019; Timurkan and Aydin, 2019; In 2013 and 2019, bovine strains were reported from Iran, also allocated as BVDV-1a, but showing atypical sequences. Based on the issues arising from genotype classification, a wider evaluation procedure, not limited to primary sequence analysis, but extended to the application of the palindromic nucleotide substitutions (PNS) genotyping method was considered for the taxonomical segregation through the exclusive consideration of strategic genomic secondary structure sequences corresponding to the internal ribosome entry site (IRES), in the 5′-UTR, responsible for translational, transcriptional, and replication events in pestiviruses (Harasawa and Giangaspero, 1998; Giangaspero and Harasawa, 2007). Within pestiviruses, PNS sequence characterization easily discriminated officially defined species as well as atypical species as Giraffe, Pronghorn, or Bungowannah (Harasawa et al., 2000; Giangaspero and Harasawa, 2008, 2011) or helped to clarify erroneous classification of strains into genotypes (Giangaspero et al., 2018). In the present study, in order to revise the classification of the BVDV-1 species and determine genotypic variations in the species, the 5′-UTR genomic region of the BVDV-1 strains reported as 1l, 1v, and 1w have been analyzed with the PNS procedure. Material and MethodsStrain sequencesIn order to determine genotypic variations in the BVDV-1 species, the 5′-UTR genomic region nucleotide sequences (derived from Sanger dideoxy sequencing technology) of 149 Pestivirus strains, have been analyzed for numerical taxonomy, considering the 3 variable loci at the level of the 5′-UTR, according to the PNS genotyping method, and focusing comparison of sequence characteristics between bovine strains recently reported from China and Turkey with the others. The virus nucleotide sequences, with different geographical origin from different host species or contaminants of biological products, were obtained from the DDBL/EMBL/GenBank DNA database (Table 1). The majority of the tested virus sequences originated from strains isolated from cattle (Bos taurus) from China (n=29) (Deng et al., 2020; Shi et al., 2020; Tian et al., 2021; Zhu et al., 2022; from Turkey (n=25) (Oguzoglu et al., 2019; Timurkan and Aydin, 2019; and from Iran (n=5). In addition, two strains were isolated in small ruminants (Capra hircus) from China (Mao et al., 2016). BVDV-1 species strains (n=88), reference for the different genotypes reported in the species, previously analyzed by the PNS method (Giangaspero and Apicella, 2014; Giangaspero et al., 2018, 2019b) have also been evaluated in the present study, in order to verify the allocation in the BVDV-1 species. Secondary structure analysisQualitative and quantitative evaluation of genomic sequence divergence, in terms of palindromic nucleotide base pairings variations, has been applied for taxonomical segregation, through the evaluation of relevant secondary structure regions in the 5′-UTR of the viral RNA, the three variable regions, V1, V2, and V3 genomic sequences, according to the genotyping based on the PNS method (Harasawa and Giangaspero, 1998; Giangaspero and Harasawa, 2007). Secondary structures were obtained for the entire 5′-UTR sequence of each strain. Palindromic sequences corresponding to the IRES three variable loci were identified in the predicted secondary structure and considered out of the rest of the nucleotide sequence. Nucleotide sequence secondary structures were predicted according to the algorithm of Zuker and Stiegler (1981) using the Genetyx-Mac version 10.1 program package (Software Development Co., Ltd., Tokyo, Japan). The minimum free energy was calculated by the method of Freier et al. (1986). The PNS software version 2.0 (Giangaspero and Apicella, 2014), prepared for the application on the genotyping procedures with the keys for Pestivirus identification of genomic sequences, using the C# programming language, was also applied for the construction of secondary structure sequence alignment, in order to compute genetic distance among strains. Segregation of BVDV-1 species strains into genotypes and relatedness among genotypes within the species was evaluated according to changes in nucleotide bps at the level of the secondary palindromic structure of the three variable loci. Genotypes were identified according to bp combinations at the level of low-variable positions (LVPs), and ranked according to increasing divergence in the genus (group reference strain value), with reference to prevalent bps in prevalent positions, the most common sequences observed, constituting the homogeneous core group within the genus. Thus, the numeric order of the nomenclature reflected genotypes’ level of divergence within the species. Among genotypes, homology was evaluated in terms of shared bps in the three variable loci in the 5′-UTR. Cross comparison between groups within the genus has been evaluated by computing the divergence percentage, identifying strains showing multiple relation (sequences sharing base pairings specific to different genomic groups, and scoring low divergence values) or borderlines (sequences showing qualitative similarities with a genomic group, but with high divergence values, candidates for reallocation as separate groups in the genus), and indicating divergence within groups and among groups quantifying the heterogeneity of a genotype and the genetic distance between groups. Table 1. List of BVDV-1 species genotypes strains (n 149) evaluated according to palindromic secondary structure characteristics at the RNA 5′-UTR (PNS method).
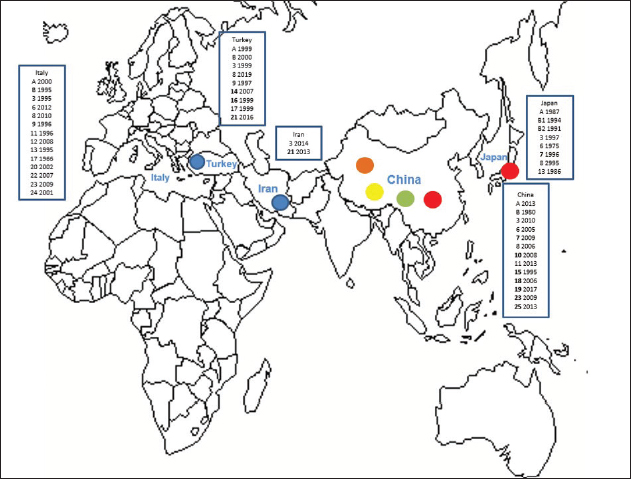
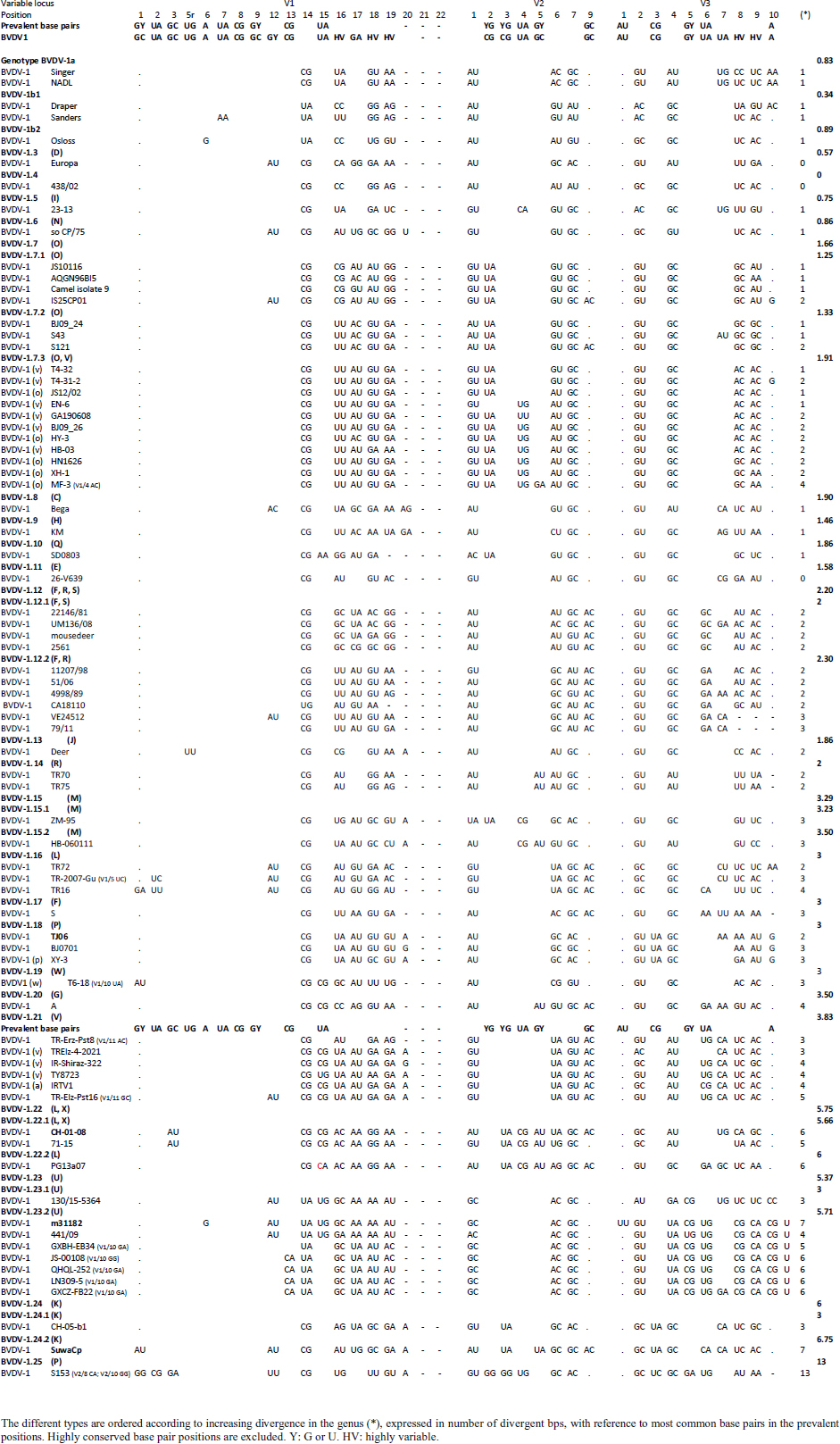
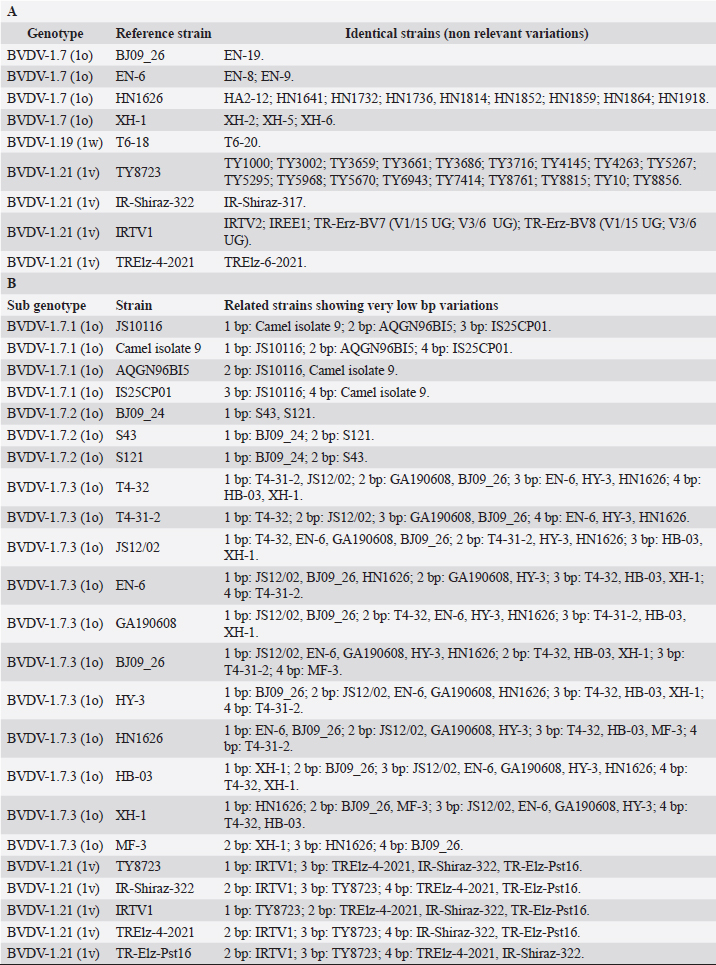
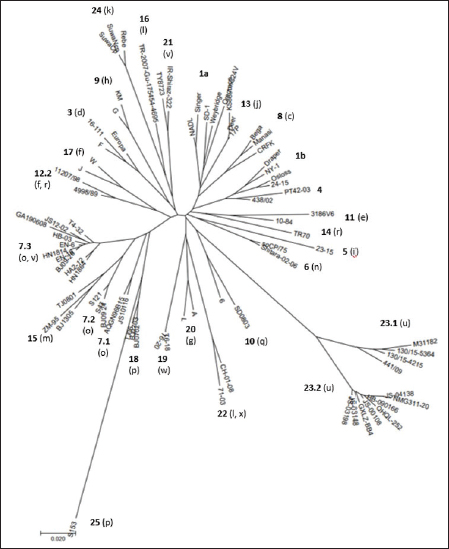
Primary sequence analysisA phylogenetic tree based on the 5′-UTR was constructed following sequence alignment with Clustal X (Thompson et al., 1997) by using the neighbor-joining method (Saitou and Nei, 1987). In addition, a basic local alignment search tool (BLAST; http://www.ncbi.nlm.nih.gov) web-based sequence analysis tool with default values was used to find homologous hits for the 5′-UTR sequence analysis of the considered strains, reported as genotypes v and w. The classification among BVDV-1 strains according to the PNS analysis based on changes in the secondary structure was compared with those based on primary structure of the 5′-UTR performed through sequence alignment and construction of phylogenetic trees by all other authors reporters of the strains considered in the present study and named according the nomenclature proposed by different authors as Vilček et al. (2001), with 11 genotypes, and subsequent further application by others authors. Ethical approvalThe local ethics committee was consulted and deemed full ethical approval unnecessary. ResultsThe observation made on the nucleotide sequences allowed the identification of consensus motifs shared by all the Pestivirus species, genus-specific base-pairings (10 PNS positioned in the V1 and V2 loci), and characteristic species and genotype-specific PNS. Based on the divergence limit value of 9 bp for genotype determination (Giangaspero and Harasawa, 2007), 25 genotypes within the BVDV-1 species have been identified, from 1a to 1.25. The geographical distribution of the newly described atypical genotypes BVDV-1.19 and BVDV-1.21 is presented in Figure 1, with the list of all other genotypes reported in the concerned countries (including Italy and Japan for comparison), and the year of collection. Secondary sequence construction, efficiently obtained by both available software, Genetyx and PNS (Giangaspero and Apicella, 2014), revealed a conserved palindromic structure in the species. Different base pairing combinations were identified for genotype characterization and considered for identification marker definition. The predicted secondary structures of the three variable loci were aligned for comparison of base pairings in the different positions (Table 2). Strains showing sequence identity at the level of the three variable loci or sharing non-relevant variations as G*U or G-C (G:Y) were excluded (Table 3). Some strains showed very low bp variations, indicating close genetic relatedness (Table 3). At the species level, the observed taxonomic status of the examined strains corresponded to the estimation obtained by phylogenetic trees constructed from the alignment with the representative strains from the identified genogroups (Fig. 2).
Fig. 1. Geographic distribution of the atypical genotypes of the BVDV-1 species 1.19 (1w) (brown circle), 1.21 (1v) (blue circle), and 1.7 sub genotypes 7.1, 7.2, and 7.3 (red, green, and yellow circles, respectively). The other genotypes reported from Turkey, Iran, and China are listed with the year of first isolation. Studies on BVDV in Iran were mainly based on seroprevalence, not completed by genetic characterization of isolates. Data were compared to the occurrence of the different genotypes reported in Italy and Japan. BVDV-1 genotypes 1.4 and 1.5 were reported as 1b and 1i only in UK, Spain, Portugal, and Egypt. Table 2. Alignment of BVDV-1 species genotypes variable loci 5’-UTR RNA secondary structure sequences, segregated according to types of bp combinations.
Table 3. BVDV-1 species strains showing sequence identity (A) and showing very low bp variations (B) at the level of the three variable loci.
Table 4. Reorganization of genotype BVDV-1.7 (1o): the determinative LVP genotype markers (V1/14 C-G, V2/7 G:Y and V3/4 G-C), positions with shared common bp (V2/2 U-A and V3/2 G*U) and exceptions in V1/4 and 12, V2/5 and 9, and V3/7 and 10 were excluded in order to consider only specific qualitative differences to characterize subgroups and verify homogeneity.
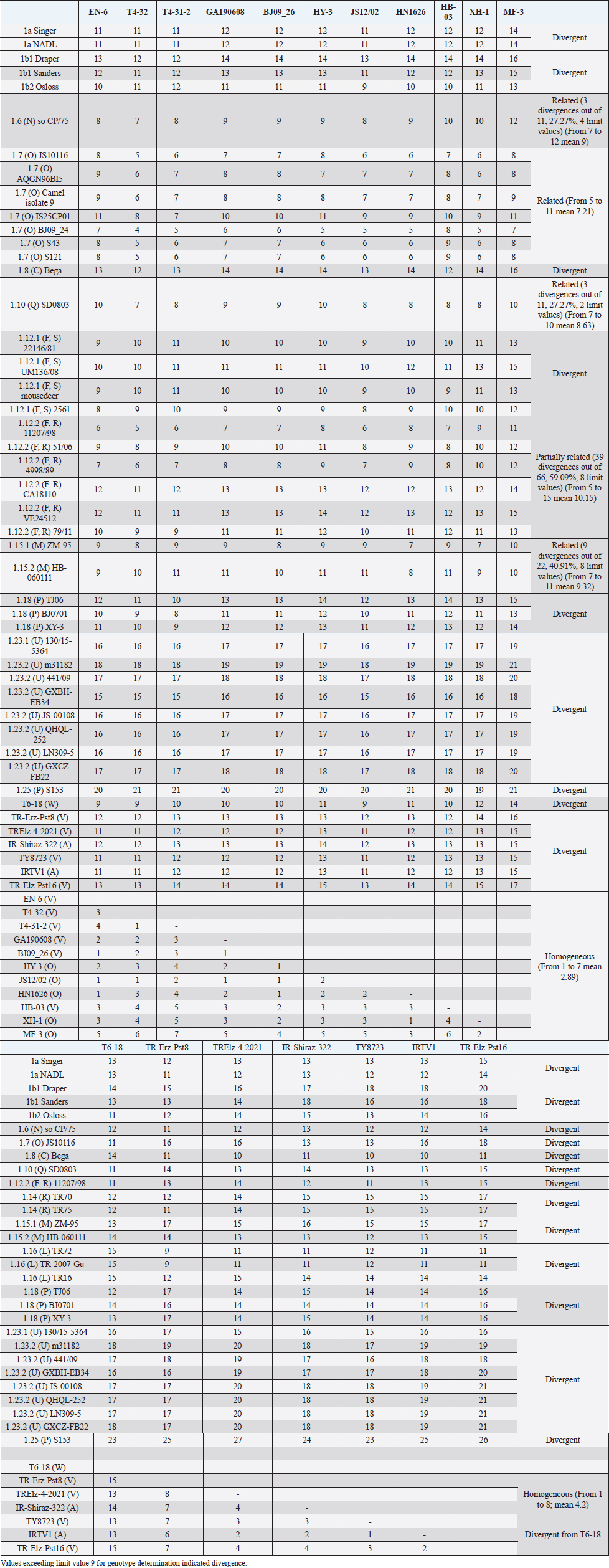
The secondary structure bps combinations of the 11 Chinese strains EN-6, T4-32, T4-31-2, GA190608, BJ09_26, HB-03 (reported as 1v), and JS12/02, HN1626, HY-3, XH-1, and MF-3 (reported as 1o) constituted a homogeneous group (divergence values ranging from 1 to 7, mean 2.89). Similarly, the Turkish strains TR-Erz-Pst8, TRElz-4-2021, TY8723, and TR-Elz-Pst16, reported as 1v, and the Iranian strains IR-Shiraz-322 and IRTV1, also reported as 1a, constituted a homogeneous group (divergence values from 1 to 8; mean 4.2). The Chinese strains T6-18 and T6-20 (reported as 1w) were divergent from all other BVDV-1 sequence clusters. No relationship among these three groups could be observed. The considered strains reported as 1v or 1o originated from China were divergent from the Turkish and Iranian 1v strains (deposited as 1a or 1l), with divergence values from 11 to 17, and divergent from the Chinese strains reported as 1w, with values from 9 to 14. Similarly, the 1w Chinese strains were divergent from the Turkish and Iranian strains with divergence values from 13 to 15. Comparison of Chinese strains reported as 1o or 1v (EN-6, T4-32, T4-31-2, GA190608, BJ09_26, HB-03, JS12/02, HN1626, XH-1, HY-3, and MF-3) with other representatives of the BVDV-1 species showed divergence from the majority of the genotypes. Distance from classical BVDV-1 genotypes 1a and 1b exceeded the limit value, ranging from 10 up to 16 divergent bp. In particular, the Chinese strains were divergent also from other Asian genotypes 1.8 (1c), 1.18 (1p), 1.23 (1u), and 1.25. Divergence from strain Bega of genotype BVDV-1.8 ranged from 12 to 16 bp, from strains of genotype 1.18 from 8 to 15, and from strains of genotype 1.23 from 15 to 20 bp. High divergence was also obtained with strain S153 (genotype 1.25) with values from 19 to 21 bp. However, they were related to the Asian genotypes 1.10 (1q) (divergence 27.27%, values from 7 to 10, mean 8.63), genotype 1.6 (1n) (divergence 27.27%, values from 7 to 12, mean 9), genotype 1.15 (1m) (divergence 40.91%, value from 7 to 11, mean 9.32), and with the highest relatedness with genotype 1.7 (1o) (divergence 7.79%, only 6 divergences out of 77 comparisons, values from 5 to 11, mean 7.21). The group was also partially related with sub genotype 1.12.2 (1f or 1r) (divergence 59.09%, values from 5 to 15, mean 10.15). The partial relation was particularly evident with the bovine German strain 11207/98, reference in the sub genotype 1.12.2, showing very low divergence values, from 5 to 8. But the group was also divergent from sub genotype 1.12.1 (1f or 1s) (divergence 72.73%, values from 8 to 15, mean 10.29). The genotype 1.7 (1o) was subdivided into three sub-genotypes (divergence limit value of 6 bp for sub-genotype determination): 7.1, 7.2, and 7.3 numbered according to their divergence in the species, 1.25, 1.33, and 2.82 divergence value mean, respectively. In subgenotype BVDV-1.7.1 were allocated four strains: JS10116, AQGN96BI5, Camel isolate 9, IS26/01ncp, and IS25CP01. In subgenotype 1.7.2 were clustered strains BJ09_24, S43, and S121. Subgenotype BVDV-1.7.3 included the strains JS12/02, HN1626, XH-1, HY-3, and MF-3, reported as 1o. Strain MF-3 showed an adenine cytosine bulge at the level of V1/4, a particular exception of the genus marker guanine cytosine or uracil (G:Y) base pairing (Giangaspero and Apicella, 2018). The strains T4-32, T4-31-2, EN-6, GA190608, HB-03, and BJ09_26, reported as 1v, were also included in subgenotype 1.7.3. All BVDV-1.7 (1o) strains shared the determinative LVP root C-G, G:Y, G-C in V1/14, V2/7, and V3/4, respectively. At the level of additional markers (V2/2 U-A; V3/9 A-U/A A/G-C), in V2/2, an exception was made for strain EN-6 which showed a typical BVDV-1 C-G, all other strains showed the characteristic 1.7 (1o) U-A base pairing (shared only with Chinese genotypes 1.10 and 1.15—1q and 1m). In V3/9, 9 strains, out of the 11 new considered strains, showed an A C bulge, different from previously described A-U/A A/G-C bp. New differences were at the level of positions V2/6 (A-U in nine strains, instead of G*U) and V3/8 (A C bulge in eight strains, instead of G-C). The allocation of the strains in the genotype BVDV-1.7 (1o) was reorganized. The determinative LVP genotype markers (V1/14 C-G, V2/7 G:Y, and V3/4 G-C), positions with shared common bp (V2/2 U-A and V3/2 G*U), and exceptions in V1/4 and 12, V2/5 and 9, and V3/7 and 10 were excluded in order to consider only specific qualitative differences to characterize sub groups and verify homogeneity. Base pairing divergence values exceeding the limit of sub-genotype determination (six divergent bps) were considered for computing clustering into sub -genotypes. Based on base pairing characteristics and divergence values (qualitative and quantitative analysis), the genotype BVDV-1.7 (1o) was clustered into three different homogeneous sub genotypes (Table 4). Within the sub-genotypes, there were no differences in terms of sequence peculiarities among cattle, the predominant host species, the strain isolated from a batracian camel of 1.7.1 or the two strains isolated from goats, belonging to 1.7.3. A comparison of Chinese strains reported as 1w (T6-18 and T6-20) showed divergence from the all the other genotypes in the BVDV-1species. Distance from classical BVDV-1 genotypes 1a and 1b ranged from 11 to 14 divergent bp. Also, with other Asian genotypes the divergence was constantly high, from values of 11 with genotypes 1.7 (1o) and 1.10 (1q), 12 with genotype 1.6 (1n), 12–14 with genotype 1.18 (1p), 14 with genotype 1.8 (1c), 13–14 with genotype 1.15 (1m), and highest values of 16–18 with genotype 1.23 (1u) and 23 divergent pb with genotype 1.25. The considered Turkish and Iranian strains were also divergent from all the other clusters in the BVDV-1 species. The divergence with reference strains of genotype 1a was 12–13 bp and with 1b ranged from 13 to 18. When compared with other Asian genotypes, the divergence was 10–16 with genotypes 1.6 (1n), 1.7 (1o), 1.8 (1c), 1.10 (1q) 1.15 (1m), and 1.18 (1p) and up to 18–24 with genotypes 1.23 (1u) and 1.25. The comparison of the Turkish and Iranian strains with atypical Turkish strains previously reported as 1.14 (1r) and 1.16 (1l) also showed divergence. With the strains TR70 and TR75 (1.14) divergence ranged from 11 to 17, mean 14.58, and with strains TR16, TR72, and TR-2007-Gu-175454-4695 (1.16) from 9 to 15, mean 11.83. At the level of PNS consensus motifs shared by all species in the genus, the strain T6-18 showed an exception in V1/10: U-A pairing instead of A-U. Similarly, in the V1 stem position 11, the strains TR-Erz-Pst8 and TR-Elz-Pst16 were characterized by an A C bulge and a G-C pairing, respectively, instead of the conserved bulge formed by two cytosine nucleotides.
Fig. 2. Phylogenetic tree based on the 5′-UTR comparison, suggesting a taxonomic position of the BVDV-1 strains in the genus Pestivirus. Strain NADL [M31182] is the reference for the BVDV-1a genotype, strains Draper [L32880] and Osloss [M96687] are the references for the BVDV-1b genotype, sub genotypes 1b1 and 1b2. Strains Europa [AB000898], 438/02 [AY159540], 23-15 [AF298059], and so CP/75 [AB042661] are references for the BVDV-1.3 to BVDV-1.6 genotypes. Strains JS10116 [JN248734], BJ09_24 [HQ116550] and BJ09-26 [HQ116551] are reference for the three sub genotypes of the BVDV-1.7 genotype, circulating exclusively in China. CRFK [D50814], KM [AF298068], SD0803 [JN400273], 10-84 [AF298054], 11207/98 [AJ304390], 17P [AF244954], TR70 [MG670547], ZM-95 [AF526381], TR-2007-Gu-175454-4695 [EU716150], J [AF298067], and TJ06 [GU120246] are references for the BVDV-1.8 to BVDV-1.18 genotypes, and strain A [AF298064] is reference for the genotype BVDV-1.20. Strains T6-18 [MN417892] and TY8723 [MH673456] are references for the BVDV-1.19 and BVDV-1.21 genotypes, restricted to China and Turkey, and Iran, respectively. Strains CH-01-08 [EU180024], M31182 (Yak) [JQ799141], Rebe [AF299317], and S153 [KF006964] are references for the BVDV-1.22 to BVDV-1.25 genotypes. Scale bar indicates 10 nucleotide substitutions per 100 nucleotides. Nomenclature of identified genotypes is based on divergence in the genus. Clustering according to primary structure analysis by depositors is indicated under parenthesis. The evaluation of the sequences by BLAST showed a similar order of genetic distance observed by the alignment of a sequence secondary structure. A low percentage of nucleotide identity was obtained when comparing the Chinese strains reported as 1v with BVDV-1b2 strains (89.52%). Higher identity was observed by comparing the Chinese strains reported as 1v with other Chinese strains reported as 1o. The strain HY-3 (1o) shared 98% identities with T4-31-2 (1v). The strain EN-6 (1v) was 99.59% identical with strain BJ09_26 (1v) and 98.78% with strains HN1732, HN1736, HN1814, and HN1859, all reported as 1o, 98.59% with T4-32 (1v) and the strains 1o HA2-12, HY-3, and XH-6, 97.88% with JS12/02 (1o) and 97.52% with strains XH-1, XH-2, XH-5, and MF-3 (1o). Similarly, the BVDV-1 strain IR-Shiraz-322 [LC053995], reported from Iran, showed 97% identities with the Turkish strain TY8723, deposited as 1l and reported as 1v. Taking into account the mean divergence values in the Pestivirus genus of the characterized groups (3 and 3.83 for the strains 1w from China and 1v, 1l, or 1a from Turkey and Iran, respectively) and that the PNS nomenclature is based on the increasing divergence in the genus, the nomenclature was modified accordingly. The group of the Chinese strains T6-18 and T16-20 (1w) was clustered in the species as BVDV-1.19 and the group from Turkey and Iran (1v, 1l, or 1a) of strains TR-Erz-Pst8, TY8723, IR-Shiraz-322, TRElz-4-2021, IRTV1, and TR-Elz-Pst16 as BVDV-1.21. Subsequently, the genotype BVDV-1.19 (1g), with a mean divergence value in the genus of 3.5 bp, was renamed 1.20, and the genotypes BVDV-1.20 (1l or x), BVDV-1.21 (1u), BVDV-1.22 (1k), and BVDV-1.23, with a divergence in the genus higher than 5 bp, were renamed as 1.22, 1.23, 1.24, and 1.25. Details of the computing of divergence values (exceeding limit value 9 for genotype determination) based on 5′-UTR secondary structure sequence alignment of BVDV-1 bovine strains EN-6, T4-32, T4-31-2, GA190608, BJ09_26, HY-3, and HN1626, HB-03, XH-1, MF-3 and one goat strain JS12/02, reported as genotype 1o or 1v isolated from China (Mao et al., 2016; Deng et al., 2020; Shi et al., 2020; Tian et al., 2021; Zhu et al., 2022; strain T6-18, reported as genotype 1w isolated from China (Deng et al., 2020), strains TY8723, TR-Erz-Pst8, TRElz-4-2021, TR-Elz-Pst16, IRTV1, and IR-Shiraz-322, reported as genotype 1a, 1l, or 1v, isolated from Turkey and Iran (Oguzoglu et al., 2019; Timurkan and Aydin, 2019; clustered according to PNS method as 1.7.3, 1.19, and 1.21, respectively are presented in Supplementary Material 1. DiscussionTaxonomyThe taxonomy of the Pestivirus A, BVDV-1, species was updated and clarified, in particular with concern to the strains of Chinese and Turkish origin, both reported as genotype 1v. In order to verify their allocation in the genus, sequences have been compared with other Pestivirus species strains previously analyzed by the PNS method (Giangaspero and Apicella, 2014; Giangaspero et al., 2018, 2019b), reaching a total number of 1,487 observations obtained with PNS procedure. Divergence values were used as reference for genotype and sub genotype allocation of the considered strains (divergence limit value of 9 and 6 bps, respectively). For example, the goat strain XY-3, reported as BVDV-1p (Mao et al., 2016), diverged only 3 bp from the strain BJ0701, reference strain of the genotype 1.18 (1p) or the Chinese camel isolate 9, clustered as 1m (Gao et al., 2013), diverged of only 2 bp from the strain AQGN96BI5 (Yamamoto et al., 2008) of genotype 1.7 (1o), and 4 bp from the 1.7 reference strain IS25CP/01 (Nagai et al., 2008), thus justifying the reallocation in this BVDV group. The Turkish strains considered in the present study were reported as 1l (Timurkan and Aydın, 2019) or 1v (Oguzoglu et al., 2019; The strains reported as 1v by Oguzoglu et al. (2019), as the bovine strain TY8856, first they have been deposited as genotype BVDV-1l, a genotype considered as the predominant sub-type of BVDV-1 in Eastern Anatolia, Turkey, based on the phylogenetic analysis of 5′-UTR, Npro, and E2 genomic regions of isolates from cattle (Timurkan and Aydın, 2019). The 5′-UTR, complete E2 gene and Npro gene region sequences of these strains have been deposited under accession numbers from MH673456 to MH673474 for 5′-UTR, from MH673439 to MH67344, and from MH67346 to MH673455 for E2, and from MH758720 to MH758737 for Npro, respectively. Erroneously, accession numbers of deposited sequences in GenBank of the Turkish BVDV-1v strains were indicated by Oguzoglu et al. (2019) for 5′-UTR MH673439-MH673455 and for E2 gene MH673456-MH673474. In reality, accession numbers were inverted and MH67345 was not found. The fact that authors deposited these strains as 1l indicates the difficulty to cluster atypical sequences within a heterogeneous species like BVDV-1. The divergence from other reported genetic groups induced first to refer to previous types characteristic of Turkey, like the strain TR-2007-Gu-175454-4695 (Oguzoglu et al., 2012). The same difficulty was encountered by the authors that deposited as BVDV-1a the Iranian strains IRTV1, IRTV2, and IREE1 since available software as Clustal X provides results of sequence comparison depending on considered reference strains loaded by the users. To avoid interpretation difficulties, it is, therefore, useful to accurately select reference strains and possibly apply other evaluation methods. BLAST could easily reveal a high homology of these Iranian strains with the Turkish atypical strains deposited as 1l. On the other side, the erroneous allocation of the strains was also probably due to the determinative LVP (V1/14; V2/7; V3/4) root Y:G, G:Y, and A-U shared with genotype 1a. However, observing the secondary structure by the PNS method, both group of strains from Turkey and Iran were well correlated and formed a single BVDV-1 genetic group, while clearly distant from the Chinese strains, despite the 1v homonymy. These strains have been defined as genotype BVDV-1.21, according to the PNS nomenclature, based on increasing divergence in the genus Pestivirus and thus, indicating a consistent genetic distance from the classical types as BVDV-1a and BVDV-1b, characterized by most common sequence traits, and expression of genetic evolution within the species and geographic segregation, circulating only in Anatolia and Iran, and to date not reported elsewhere. While genotype 1.19 (1w) constituted a separate group in the species, reported in China more recently in 2017 (Deng et al., 2020), the Chinese group of strains reported as 1v (Deng et al., 2020; Tian et al., 2021; Zhu et al., 2022) and those reported as 1o and considered as 1v (Shi et al., 2020; was related to genotype 1.7 (1o) and constituting a new sub genotype (1.7.3). This corresponded to the observation made by Shi et al. (2020), who reported nine Pestivirus A isolates as putative new 1o sub genotype, based on phylogenetic analysis of partial 5′-UTR and Npro sequences. The sub genotype 1.7.1 included one Chinese pig strain JS10116, reported as genotype 1o by Deng et al. (2012), the Chinese camel isolate 9, clustered as 1m (Gao et al., 2013) and three cattle isolates from Japan, reported as 1o, AQGN96BI5 (Yamamoto et al., 2008), IS26/01ncp and IS25CP/01 (Nagai et al., 2008). Three Chinese strains, all reported as genotype 1m, from cattle, S43, S121, and BJ09_24 (Zhang et al., 2014b) were clustered in the sub-genotype BVDV-1.7.2. Sub genotypes 1.7.2 and 1.7.3 diverged from 1.7.1 at the level of V1 locus in positions 16, 18, and 19 with U U bulge, G*U or G A and G A or A A bulges, respectively (Table 4). In addition, 1.7.2 showed characteristic bp A-U in V2/1 and G-C in V3/9. Turkey and IranSince the first report of BVDV in Turkey in the 1990’, in addition to genotype BVDV-1.21, other rare BVDV-1 strains were characteristic of the country and expression of geographic segregation: the BVDV-1.16 bovine strains reported as genotype 1l (Oguzoglu et al., 2012), and strains TR70, TR73 and TR75 belonging to the BVDV-1.14, reported as genotype 1r, restricted to Turkey (Yesilbag et al., 2014, 2017; Giangaspero et al., 2018). Until now, nine genotypes were described in Turkey. In addition to the atypical genetic clusters, other classical genotypes have been reported: 1a and 1b, 1.3 (d), 8 (c), 9 (h), and 17 (f). In Iran, BVDV was first described by Mirshamsy et al. (1970). Studies were mainly based on serology showing a high prevalence of BVDV antibodies in cattle in some areas of the country, up to 100% (Kargar Moakhar et al., 1995; Khezri, 2015). However, studies were generally not completed by sequencing and characterization of isolates, and only a few reports and deposited sequences are available (Khodakaram-Tafti et al., 2016). ChinaThe first report of BVDV in China was in the north eastern province of Jilin in 1980 related to 1b infected cattle imported from Europe (Li et al., 1983). The phylogenetic analysis clustered the first BVDV strain CC-184, which was isolated in China from the cows imported from Europe, to BVDV-1b (Li et al., 1983; Wang et al., 1996). The majority of BVDV strains isolated later from Jilin and other regions in China, including isolates JL-1 [KF501393] in 2009 and BVDV-CC13B [KF772785] in 2013, were of genotype BVDV-1b. These strains shared the highest sequence identity with the CP7 strain [U63479], originally isolated in Germany in 1987 (Zhang et al., 2014a; Zhu et al., 2014). Those results supported the hypothesis that the strains of the predominant genotype BVDV-1b currently spread in China were originally from European countries (Shi and Zhang, 1987; Wang et al., 1996; Zhu et al., 2016), and probably from Germany. Similarly, in the present study, genetic characteristics suggested the evolution of the virus population in China from strains introduced from Germany. The sub-genotype 1.7.3, including exclusively Chinese strains reported as 1v or 1o, was partially related to sub-genotype 1.12.2, showing an overall divergence of 59.09% (39 divergences from 5 to 15, with mean divergence value 10.15, out of 66 comparisons and 8 limit values—9 bp). The Chinese strain BJ09-26 was the first isolated of the sub genotype 1.7.3, in 2009. The other 1.7.3 strains were reported in following years from 2013 to 2019. The strains belonging to 1.12.2, reported as 1f or 1r, included the strains 11207/98 and 4998/89, isolated in Germany in 1989. The other members of the sub-genotype were strain 51/06 isolated in Poland in 2006, and the strains CA18110, 79/11, and VE24512, isolated in Italy later (from 2010 to 2012). The relationship between the two sub genotypes was particularly evident with strain 11207/98, distant only 5 bp from strain T4-32 or 6 bp from strains EN-6, T4-31-2, and JS12/02. 11207/98 was divergent of 9 bp from both strains JS10116 and BJ09_24, isolated in China in 2010 and 2009, belonging to sub genotypes 1.7.1 and 1.7.2, respectively. Taking into account the dates and places of isolation, it is likely that the origin of sub-genotype 1.7.3 is Germany, where the sub-genotype 1.12.2 appeared and diffused later in other European countries and China. Since the first introduction, BVD outbreaks were not often reported until the later 1990s and early 2000s. Further, BVD outbreaks were reported in many regions across China and over the half of the 25 genotypes described in the species, 13 could be identified: BVDV-1a, 1b, 1.3, 1.6, 1.7, 1.8, 1.10, 1.11, 1.15, 1.18, 1.19, 1.23, and 1.25. The predominant genotypes were BVDV-1b and BVDV-1.15 (1m) (Xue et al., 2010, Zhong et al., 2011). Italy and JapanThe comparison of the epidemiological situation of BVDV in China to the occurrence of the different genotypes reported in Italy showed a similar heterogeneity of the species, with specific differences at the level of circulating types. Fourteen genotypes were observed in Italy: BVDV-1a, 1b1, and 1b2, 1.3, 1.6, 1.8, 1.9, 1.11, 1.12, 1.13, 1.17, 1.20, 1.22, 1.23, and 1.24. The BVDV-1b and 1.11 (1e) genotypes were predominant. The occurrence of genotype 1a was also demonstrated and reported for the first time in north, central, and island (Sardinia) regions (Giammarioli et al., 2008) and then in southern regions (Giangaspero et al., 2019a). Three uncommon genotypes were reported in north and central Italy, 1.13 (j), 1.17 (f), and 1.22 (l, x) (Falcone et al., 2003; Luzzago et al., 2014; Cerutti et al., 2016). Other strains belonged to genotypes circulating in a few countries. For example, genotype 1.9 (h) was spread only in Switzerland, Austria, and Italy, and rarely reported also in UK, Slovakia, and South Africa. Genotype 1.20 (g) was rare and reported only in Austria, UK, and South Africa. The first evidence for the circulation of this genotype in Italy was reported by Giammarioli et al. (2008) and Luzzago et al. (2014), and further rarely detected also in southern Italy. Only two strains were reported, one from Basilicata (Luzzago et al., 2014) and another from Calabria (Decaro et al., 2016). Similarly, genotype 1.24 (k) was rare and reported only in Switzerland, and detected for the first time in northern and southern Italy (Luzzago et al., 2014). The few BVDV-1.6 (n), 1.8 (c), and 1.23 (u) strains detected in Italy, represented an exception in genotypes characteristic from Asia. The progressive increasing of genetic variability of BVDV-1 in Italy (3 genotypes in 2002, 7 in 2008, 12 in 2014, and 14 reported in 2019) was probably mainly due to import of live animals. More than 1,300,000 bovines are imported yearly, mainly from France and largely destined to the northern regions of Veneto, Lombardia, and Piedmont. The fact that in France BVDV-1.11 (e) is prevalent (Jackova et al., 2008), probably explains the high prevalence of this genotype in northern regions, in particular Piedmont (Luzzago et al., 2014). The introduction of BVDV-1.23 in Italy was suspected to be due to contamination of biological products for veterinary use (Giangaspero et al., 2019a). In Japan, the vaccine against BVDV was introduced since 1973, but the use was infrequent, due to the accidental production of persistently infected animals occurred following the vaccination of pregnant cows with the modified live virus. The number of BVD cases increased in recent years, with a mean of 198 cases per year (Tajima, 2021). There is currently no national eradication program for BVD in Japan, but regional voluntary eradication trials have been performed since the 2000's, along with regional surveillance to identify persistently infected cattle (Abe et al., 2016, Akagami et al., 2020). In terms of genetic diversity, BVDV may not vary to the same extent as in European countries (Nagai et al., 2001; Tajima, 2006; Abe et al., 2016). BVDV-1 species was far less heterogeneous than in China, showing seven genotypes: 1a, 1b (b1 and b2), 1.3, 1.6, 1.7, 1.8, and 1.13. However, Japan shared with China three genotypes (1.6, 1.7, 1.8) restricted to Far East Asia and Austral Asia (1.6 present also in South Korea and 1.8 also in Australia). In particular, the sub genotype 7.1 was present exclusively in Japan and China. Taking into account the earlier reporting of the variant in Japan (1996) and more than a decade later in China in 2009, this may suggest the origin of the introduction in China of a new genotype and a subsequent evolution in two additional sub types (7.2 and 7.3). Geographic segregationThe BVDV-1 species was the most heterogeneous within the genus Pestivirus, and this may be due to geographic segregation. Different BVDV-1 species genotypes, showing particular genetic characteristics, were restricted to specific geographic areas also in Asia. Some genetic variants appeared to be restricted in certain areas, as certain genotypes circulating only in Turkey or China, suggesting geographic isolation (Xue et al., 2010). Some sub genotypes and genotypes were characteristic from Asia. Sub genotypes 1.7.2 and 1.7.3 and genotypes 1.10, 1.15, 1.18, and 1.25 were circulating exclusively in China, and also genotype 1.23 has been reported almost exclusively in China. Other genotypes (1.6, 1.7.1, and 1.8) appear to be restricted to Asian or Austral Asian countries. In rare occasions, strains belonging to Asian genotypes 1.6, 1.8, and 1.23 were also reported in Italy, representing exceptions. Observing the secondary structure, the geographic segregation was associated to specific sequence characteristics. For example, Chinese strains of the genotype BVDV-1.10 showed divergence in V1/15 with A A or C A bulges, instead of BVDV-1 species marker U-A pairing. In sub genotype 1.15.2, all strains showed species marker exception in V2/5, with A-U instead of G-C. Similarly, strain S153 (Zhang et al., 2014b), genotype 1.25, showed an atypical G A bulge at the level of BVDV-1 species marker position in V3/5. The Asian clusters 1.8 and 1.15.2 shared a root characteristic of genotype BVDV-1a (V1/14 C-G, V2/7 G-C, and V3/4 A-U). All the strains belonging to group 1.18 (bovine or contaminant strains from Australia, China, and Japan) showed an A C bulge (enlargement in the RNA secondary structure) in position 12 in V1 locus, a base pairing not present in any member in the genotype 1a. Among Chinese strains of the sub-genotype 15.2, characterized also by an exception at the level of species marker in V2/5 (A-U instead of G-C), root A was present in the majority of the strains. A peculiar U-A pairing in V2/2 was present only among certain Asian genotypes (1.7, 1.10, and 1.15.1). Genotype 1.23 showed a new and atypical V1/14 U-A, V2/7 G-C, and V3/4 U-A root associated with genotype b (sub genotype 2). The V1/14 U-A was shared exclusively with BVDV-1b and BVDV-1.21 which was characteristic, and in all the BVDV-2 species strains. With reference to Asia, also in other Pestivirus species geographic segregation was observed. In the Pestivirus B (Bovine viral diarrhea virus type 2, BVDV-2) species, only one group appeared specific to Asia. The genotype b variant 4 (BVDV-2b4) included only Chinese isolates: the bovine strain SD-1301 (Wang et al., 2014) and the contaminants S143, S172, and S51 (Zhang et al., 2014b). Pestivirus H (Bovine viral diarrhea virus type 3, BVDV-3) species genotypes BVDV-3.2, BVDV-3.3, and BVDV-3.4 were specific to zebu and bovine isolates from India and Bangladesh, respectively (Haider et al., 2014; Mishra et al., 2014). Pestivirus D (Border disease virus, BDV) species sequence characteristics of Chinese and Turkish strains were highly divergent from other genogroups, indicating geographic segregation. Chinese strains, reported as genotype BDV-3 (Ghiforn type—PNS BDV-j), AH12-01, AH12-02, and AHHX15 (Li et al., 2013) have been clustered as genotype BDV-d, sub genotype d1. These strains showed high homology with strain 297 (Leskova et al., 2013), clustered in the same genotype, but as separate sub genotype BDV-d2. Similarly, other Chinese strains JS12/04, JSLS12-01, and JSYZ15 (Li et al., 2013) have been clustered as genotype BDV-h. Turkish strains TR-13 and TR-14, reported as a distinct group in the BDV species (Toplu et al., 2012), have been clustered as genotype BDV-i. The ovine Turkish strains BDV/Aydin/04-TR and BDV/Burdur/05-TR (Oguzoglu et al., 2009) represented the Pestivirus I species. In the Pestivirus C (CSFV) species, three genetic clusters referred specifically to Asian countries. The CSFV genotype a variant 4 (type Parambi) included only pig and wild boar strains from India (Bhaskar et al., 2015; CSFV pig strains clustered into genotype C (type Okinawa) were reported only from Japan and Taiwan (Harasawa and Giangaspero, 1999; Sakoda et al., 1999; Lin et al., 2007; The Chinese strain S171 (Zhang et al., 2014b), isolated from bovine serum, was clustered as CSFV-d. Potential implicationsDespite the untranslated region, by definition, it is not translated into antigens, the molecular variations at the level of 5′-UTR are correlated to parts of the BVDV genome which contribute to antigenic variation, expressing structural and nonstructural strongly immunogenic proteins as E2 and NS3 (Lazear et al., 2013; Lanyon et al., 2014; Yitagesu et al., 2021; Zheng et al., 2021; Chi et al., 2022), and thus important also for serological diagnosis and vaccine development (Mahony et al., 2015; Al-Kubati et al., 2021). Studies on antigenic similarity among genotypes, measuring virus neutralization antibody titers of hyperimmunized antiserums against studied BVDV-1 genotypes, showed the highest similarity (highest cross neutralizing antibody titers) between genotypes 1a and 1b (homologous pairs which are widely used in commercial vaccines) compared to the very weak antigenic similarity among 1a and 1b with the viruses from other different genotypes, particularly BVDV-1.17 (1f) and BVDV-1.24 (1k) (Castrucci, 1978; Bachofen et al., 2008; Alpay and Yesilbag, 2015). The growing number of reports on BVDV-1 heterogeneity raises significant concerns about the emergence and spread of new BVDV variants, with possible implications on animal health and disease control (Giammarioli et al., 2015). Although there is no clear indication that atypical strains within the species may negatively influence diagnostic analysis or impair the efficacy of the available vaccines, the antigenic differences limit the cross-protection among the highly divergent types of bovine pestiviruses. Such problems are evident with the Pestivirus H (Bauermann et al., 2013). Also, some reports concerned BVDV-1, showing a low level of antibody response to the BVDV-1b by BVDV-1a vaccines (Fulton et al., 2003) and some insufficient responses to protect against BVDV-1b infections (Grooms et al., 2007; Ridpath et al., 2010). Therefore, it is important to constantly monitor the evolution of the virus population for early detection of new variants, through the testing of field isolates, applying adequate genotyping methods. Furthermore, cross-protection studies should be carried out taking into account reported atypical strains and the regional epidemiological situation to determine whether future vaccines should be produced using several BVDV genotypes. ConclusionThe study demonstrated the potential utility of the PNS technique to resolve some of the BVDV genetic classification difficulties. The evaluation of secondary structure by PNS method of strains clustered as new BVDV-1 genotypes 1.19 (1w) and 1.21 (1v), characteristic of China, Turkey, and Iran, respectively. Confusion in nomenclature was clarified, allocating as sub genotype 1.7.3 (1o) the Chinese strains reported as 1v. The heterogeneity in pestiviruses has the potential to cause diagnostic and prophylactic difficulties because commonly available tests and vaccines are based on viral antigenic substrate (Bolin et al., 1985; Giammarioli et al., 2015). Recognition of the molecular characteristics of field strains is important for the control or eradication programs design, vaccine development or for specific purposes as the retracing of infection sources in case of outbreaks (Booth et al., 2013; Kuta et al., 2013). Increasing genetic heterogeneity in Pestivirus species, also due to geographic segregation, indicates a need for the application of different analytical procedures to avoid interpretation difficulties and obtain accurate genetic analysis, given the importance of epidemiological data, to preserve animal health and welfare against the spread of potentially virulent genetic clusters in naïve animal populations. Conflict of interestAuthors declare that there is no conflict of interest. Authors’ contributionM. Giangaspero and S. Zhang contributed equally to the present study. FundingScience and Technology of Jilin Province (20220202058NC). Data availabilityData available on request from the authors. ReferencesAbe, Y., Tamura, T., Torii, S., Wakamori, S., Nagai, M., Mitsuhashi, K., Mine, J., Fujimoto, Y., Nagashima, N., Yoshino, F., Sugita, Y., Nomura, T., Okamatsu, M., Kida, H. and Sakoda, Y. 2016. Genetic and antigenic characterization of bovine viral diarrhea viruses isolated from cattle in Hokkaido, Japan. J. Vet. Med. Sci. 78, 61–70. Akagami, M., Sekia, S., Kashima, Y., Yamashita, K., Oya, S., Fujii, Y., Takayasu, M., Yaguchi, Y., Suzuki, A., Ono, Y., Ouchi, Y. and Hayama, Y. 2020. Risk factors associated with the within-farm transmission of bovine viral diarrhea virus and the incidence of persistently infected cattle on dairy farms from Ibaraki prefecture of Japan. Res. Vet. Sci. 129, 187–192. Al-Kubati, A.A.G., Hussen, J., Kandeel, M., Al-Mubarak, A.I.A. and Hemida, M.G. 2021. Recent advances on the bovine viral diarrhea virus molecular pathogenesis, immune response, and vaccines development. Front. Vet. Sci. 8, 665128. Alpay, G. and Yesilbag, K. 2015. Serological relationships among subgroups in bovine viral diarrhoea virus genotype 1 (BVDV-1). Vet. Microbiol. 175(1), 1–6. Bachofen, C., Stalder, H., Braun, U., Hilbe, M., Ehrensperger, F. and Peterhans, E. 2008. Co-existence of genetically and antigenically diverse bovine viral diarrhoea viruses in an endemic situation. Vet. Microbiol. 131(1–2), 93–102. Barros, S.C., Ramos, F., Paupério, S., Thompson, G. and Fevereiro, M. 2006. Phylogenetic analysis of Portuguese bovine viral diarrhoea virus. Virus Res. 118(1–2), 192–195. Bauermann, F.V., Ridpath, J.F., Weiblen, R. and Flores, E.F. 2013. HoBi-like viruses: an emerging group of pestiviruses. J. Vet. Diagnost. Invest. 25, 6–15. Baule, C., van Vuuren, M., Lowings, J.P. and Belak, S. 1997. Genetic heterogeneity of bovine viral diarrhoea viruses isolated in Southern Africa. Virus. Res. 52(2), 205–220. Becher, P., Orlich, M., Shannon, A.D., Horner, G., Konig, M. and Thiel, H.-J. 1997. Phylogenetic analysis of pestiviruses from domestic and wild ruminants. J. Gen. Virol. 78, 1357–1366. Bhaskar, N., Ravishankar, C., Rajasekhar, R., Sumod, K., Sumithra, T.G., John, K., Mini, M., Ravindran, R., Shaji, S. and Aishwarya, J. 2015. Molecular typing and phylogenetic analysis of classical swine fever virus isolates from Kerala, India. Virus Dis. 26, 260–266. Bolin, S.R., McClurkin, A.W., Cutlip, R.C. and Coria, M.F. 1985. Response of cattle persistently infected with noncytopathic bovine viral diarrhea virus to vaccination for bovine viral diarrhea and to subsequent challenge exposure with cytopathic bovine viral diarrhea virus. Am. J. Vet. Res. 46, 2467–2470. Booth, R.E., Thomas, C.J., El-Attar, L.M., Gunn, G. and Brownlie, J. 2013. A phylogenetic analysis of bovine viral diarrhoea virus (BVDV) isolates from six different regions of the UK and links to animal movement data. Vet. Res. 44(1), 43. Castrucci, G. 1978. Togaviridae. Infezione da virus degli animali domestici. Bologna, Italy: Esculapio. Cerutti, F., Luzzago, C., Lauzi, S., Ebranati, E., Caruso, C., Masoero, L., Moreno, A., Acutis, P.L., Zehender, G. and Peletto, S. 2016. Phylogeography, phylodynamics and transmission chains of bovine viral diarrhea virus subtype 1f in Northern Italy. Infect. Genet. Evol. 45, 262–267. Chi, S., Chen, S., Jia, W., He, Y., Ren, L. and Wang, X. 2022. Non-structural proteins of bovine viral diarrhea virus. Virus Genes 58, 491–500. Ciulli, S., Battilani, M., Scagliarini, A., Ostanello, F. and Prosperi, S. 2003. Identificazione e caratterizzazione di ceppi di BVDV isolati da soggetti immunotolleranti nella provincia di Bologna. Vet. Ital. 38, 65–72. Collett, M.S., Larson, R., Gold, C., Strick, D., Anderson, D.K. and Purchio, A.F. 1988. Molecular cloning and nucleotide sequence of the Pestivirus bovine viral diarrhea virus. Virology 165, 191–199. Couvreur, B., Letellier, C., Collard, A., Quenon, P., Dehan, P., Hamers, C., Pastoret, P.P. and Kerkhofs, P. 2002. Genetic and antigenic variability in bovine viral diarrhea virus (BVDV) isolates from Belgium. Virus Res. 85(1), 17–28. Decaro, N., Lucente, M.S., Lanave, G., Gargano, P., Larocca, V., Losurdo, M., Ciambrone, L., Marino, P.A., Parisi, A., Casalinuovo, F., Buonavoglia, C. and Elia, G. 2016. Evidence for circulation of bovine viral diarrhoea virus type 2c in ruminants in southern Italy. Transbound. Emerg. Dis. 64(6), 1935–1944. De Moerlooze, L., Lecomte, C., Brown-Shimmer, S., Schmetz, D., Guiot, C., Vandenbergh, D., Allaer, D., Rossius, M., Chappuis, G., Dina, D., Renard, A. and Martial, J.A. 1993. Nucleotide sequence of the bovine viral diarrhoea virus Osloss strain: comparison with related viruses and identification of specific DNA probes in the 5 untranslated region. J. Gen. Virol. 74, 1433–1438. Deng, M., Chen, N., Guidarini, C., Xu, Z., Zhang, J., Cai, L., Yuan, S., Sun, Y. and Metcalfe, L. 2020. Prevalence and genetic diversity of bovine viral diarrhea virus in dairy herds of China. Vet. Microbiol. 242, 108565. Deng, M., Ji, S., Fei, W., Raza, S., He, C., Chen, Y., Chen, H. and Guo, A. 2015. Prevalence study and genetic typing of bovine viral diarrhea virus (BVDV) in four bovine species in China. PLoS One 10(4), e0121718. Erratum in: PLoS One 10(7), e0134777. Deng, R. and Brock, K.V. 1992. Molecular cloning and nucleotide sequence of the Pestivirus genome, non-cytopathic bovine viral diarrhea virus strain SD-1. Virology 191, 867–879. Deng, Y., Shan, T.L., Tong, W., Zheng, X.C., Guo, Y.Y., Zheng, H., Cao, S.J. and Wen, X.T. 2014. Genomic characterization of a bovine viral diarrhea virus 1 isolate from swine. Arch. Virol. 159(9), 2513–2517. Deng, Y., Sun, C.Q., Cao, S.J., Lin, T., Yuan, S.S., Zhang, H.B., Zhai, S.L., Huang, L., Shan, T.L., Zheng, H., Wen, X.T. and Tong, G.Z. 2012. High prevalence of bovine viral diarrhea virus 1 in Chinese swine herds. Vet. Microbiol. 59(3–4), 490–493. Falcone, E., Cordioli, P., Tarantino, M., Muscillo, M., La Rosa, G. And Tollis, M. 2003. Genetic heterogeneity of bovine viral diarrhoea virus in Italy. Vet. Res. Commun. 27(6), 485–494. Freier, S.M., Kierzek, R., Jaeger, J.A., Sugimoto, N., Caruthers, M.H., Nielson, T. and Turner, D.H. 1986. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. USA 83, 9373–9377. Fulton, R.W., Ridpath, J.F., Confer, A.W., Saliki, J.T., Burge, L.J. and Payton, M.E. 2003. Bovine viral diarrhoea virus antigenic diversity: impact on disease and vaccination programmes. Biologicals 31(2), 89–95. Gao, S., Du, J., Shao, J., Lang, Y., Lin, T., Cong, G., Zhao, F., Belák, S., Liu, L., Chang, H. and Yin, H. 2014. Genome analysis reveals a novel genetically divergent subgenotype of bovine viral diarrhea virus in China. Infect. Genet. Evol. 21, 489–491. Gao, S., Luo, J., Du, J., Lang, Y., Cong, G., Shao, J., Lin, T., Zhao, F., Belak, S., Liu, L., Chang, H. and Yin, H. 2013. Serological and molecular evidence for natural infection of bactrian camels with multiple subgenotypes of bovine viral diarrhea virus in western China. Vet. Microbiol. 163, 172–176. Giammarioli, M., Ceglie, L., Rossi, E., Bazzucchi, M., Casciari, C., Petrini, S. and De Mia, G.M. 2015. Increased genetic diversity of BVDV-1: recent findings and implications thereof. Virus Genes 50(1), 147–151. Giangaspero, M. and Apicella, C. 2014. Improved palindromic nucleotide substitutions software version 2.0. genotyping based on the secondary structure alignment in the 5’ untranslated region of Pestivirus RNA. J. Bioinform. Intell. Cont. 3(1), 39–64. Giangaspero, M. and Apicella, C. 2018. Bovine viral diarrhea virus type 1 current taxonomy according to palindromic nucleotide substitutions method. J. Virol. Methods 256, 37–76. Giangaspero, M., Decaro, N., Turno, P., Apicella, C., Gargano, P. and Buonavoglia, C. 2019a. Pathogen spread and globalization: the case of Pestivirus heterogeneity in southern Italy. Res. Vet. Sci. 125, 100–112. Giangaspero, M. and Harasawa, R. 2007. Numerical taxonomy of genus Pestivirus based on palindromic nucleotide substitutions in the 5’ untranslated region. J. Virol. Methods 146, 375–388. Giangaspero, M. and Harasawa, R. 2008. Genetic variation of classical swine fever virus based on palindromic nucleotide substitutions, a genetic marker in the 5′ untranslated region of RNA. Vet. Ital. 44, 305–318. Giangaspero, M. and Harasawa, R. 2011. Species characterization in the genus Pestivirus according to palindromic nucleotide substitutions in the 5′ untranslated region. J. Virol. Methods 174(1–2), 166–172. Giangaspero, M., Harasawa, R. and Verhulst, A. 1997. Genotypic characteristics of the 5’ untranslated region of a Pestivirus strain isolated from human leucocytes. Microbiol. Immunol. 40, 829–834. Giangaspero, M., Vacirca, G., Harasawa, R., Büttner, M., Panuccio, A., De Giuli Morghen, C., Zanetti, A., Belloli, A. and Verhulst, A. 2001. Genotypes of Pestivirus RNA detected in live virus vaccines for human use. J. Vet. Med. Sci. 63(7), 723–733. Giangaspero, M., Yesilbag, K. and Apicella, C. 2018. Who's who in the bovine viral diarrhea virus type 1 species: genotypes L and R. Virus Res. 256, 50–75. Giangaspero, M., Zhang, S. and Apicella, C. 2019b. Heterogeneity of Pestivirus species in Asia. Adv. Microbiol. 9, 266–342. Grondahl, C., Uttenthal, A., Houe, H., Rasmussen, T.B., Hoyer, M.J. and Larsen, L.E. 2003. Characterisation of a Pestivirus isolated from persistently infected mousedeer (Tragulus javanicus). Arch. Virol. 148(8), 1455–1463. Grooms, D.L., Bolin, S.R., Coe, P.H., Borges, R.J. and Coutu, C.E. 2007. Fetal protection against continual exposure to bovine viral diarrhea virus following administration of a vaccine containing an inactivated bovine viral diarrhea virus fraction to cattle. Am. J. Vet. Res. 68(12), 1417–1422. Haider, N., Rahman, M.S., Khan, S.U., Mikolon, A., Gurley, E.S., Osmani, M.G., Shanta, I.S., Paul, S.K., Macfarlane-Berry, L., Islam, A., Desmond, J., Epstein, J.H., Daszak, P., Azim, T., Luby, S.P., Zeidner, N. and Rahman, M.Z. 2014. Identification and epidemiology of a rare HoBi-Like Pestivirus strain in Bangladesh. Transbound. Emerg. Dis. 61, 193–198. Harasawa, R. 1996. Phylogenetic analysis of Pestivirus based on the 5’ untranslated region. Acta Virol. 40, 49–54. Harasawa, R. and Giangaspero, M. 1998. A novel method for Pestivirus genotyping based on palindromic nucleotide substitutions in the 5'-untranslated region. J. Virol. Methods 70, 225–230. Harasawa, R. and Giangaspero, M. 1999. Genetic variation in the 5’ end and NS5B regions of classical swine fever virus genome among Japanese isolates. Microbiol. Immunol. 43, 373–379. Harasawa, R., Giangaspero, M., Ibata, G. and Paton, P.J. 2000. Giraffe strain of Pestivirus. Its taxonomic status based on the 5′ untranslated region. Microbiol. Immunol. 44(11), 915–921. Harasawa, R. and Mizusawa, H. 1995. Demonstration and genotyping of Pestivirus RNA from mammalian cell lines. Microbiol. Immunol. 39, 979–985. Hurtado, A., Garcia-Perez, A.L., Aduriz, G. and Juste, R.A. 2003. Genetic diversity of ruminant pestiviruses from Spain. Virus Res. 92, 67–73. Kargar Moakhar, R., Ahuraei, P., Hesami, M., Khedmati, K., Gholami, M.R. and Kazemi, A. 1995. Reporting presence and prevalence of BVD/MD in cattle farms around Tehran. J. Pajohesh Sazandegi 28, 112–116. Khezri, M. 2015. Bovine viral diarrhea (BVD): a review emphasizing on Iran perspective. J. Adv. Vet. Anim. Res. 2(3), 240–251. Khodakaram-Tafti, A., Mohammadi, A. and Farjani Kish, G.H. 2016. Molecular characterization and phylogenetic analysis of bovine viral diarrhea virus in dairy herds of Fars province, Iran. Iran. J. Vet. Res. 17(2), 89–97. Kuta, A., Polak, M.P., Larska, M. and Zmudzinski, J.F. 2013. Predominance of bovine viral diarrhea virus 1b and 1d subtypes during eight years of survey in Poland. Vet. Microbiol. 166(3–4), 639–644. Jackova, A., Novackova, M., Pelletier, C., Audeval, C., Gueneau, E., Haffar, A., Petit, E., Rehby, L. and Vilcek, S. 2008. The extended genetic diversity of BVDV-1: typing of BVDV isolates from France. Vet. Res. Commun. 32(1), 7–11. Jones, L.R., Zandomeni, R. and Weber, E.L. 2001. Genetic typing of bovine viral diarrhea virus isolates from Argentina. Vet. Microbiol. 81(4), 367–375. Lanave, G., Decaro, N., Lucente, M.S., Guercio, A., Cavaliere, N., Purpari, G., Padalino, I., Larocca, V., Antoci, F., Marino, P.A., Buonavoglia, C. and Elia, G. 2017. Circulation of multiple subtypes of bovine viral diarrhoea virus type 1 with no evidence for HoBi-like Pestivirus in cattle herds of southern Italy. Infect. Genet. Evol. 50, 1–6. Lanyon, S.R., Hill, F.I., Reichel, M.P. and Brownlie, J. 2014. Bovine viral diarrhoea: pathogenesis and diagnosis. Vet. J. 199(2), 201–209. Lazear, H.M., Lancaster, A., Wilkins, C., Suthar, M.S., Huang, A., Vick, S.C., Clepper, L., Thackray, L., Brassil, M.M., Virgin, H.W., Nikolich-Zugich, J., Moses, A.V., Gale, M. Jr., Früh, K. and Diamond, M.S. 2013. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 9(1), e1003118. Leskova, V., Jackova, A., Vlasakova, M. and Vilcek, S. 2013. Genetic characterization of a border disease virus isolate originating from Slovakia. Acta Virol. 57, 17–25. Li, W., Mao, L., Zhao, Y., Sun, Y., He, K. and Jiang, J. 2013. Detection of border disease virus (BDV) in goat herds suffering diarrhea in Eastern China. Virol. J. 10, 80. Li, Y., Liu, Z. and Wu, Y. 1983. Isolation and identification of bovine viral diarrhea virus-mucosal disease virus strain Changchun 184. Chin. J. Vet. Sci. 3(2), 546–553. Lin, Y.J., Chien, M.S., Deng, M.C. and Huang, C.C. 2007. Complete sequence of a subgroup 3.4 strain of classical swine fever virus from Taiwan. Virus Genes 35, 737–744. Luzzago, C., Lauzi, S., Ebranati, E., Giammarioli, M., Moreno, A., Cannella, V., Masoero, L., Canelli, E., Guercio, A., Caruso, C., Ciccozzi, M., De Mia, G.M., Acutis, P.L., Zehender, G. and Peletto, S. 2014. Extended genetic diversity of bovine viral diarrhea virus and frequency of genotypes and subtypes in cattle in Italy between 1995 and 2013. Biomed. Res. Int. 2014, 147145. Mahony, D., Mody, K.T., Cavallaro, A.S., Hu, Q., Mahony, T.J., Qiao, S. and Mitter, N. 2015. Immunisation of sheep with bovine viral diarrhoea virus, E2 protein using a freeze-dried hollow silica mesoporous nanoparticle formulation. PLoS One 10(11), e0141870. Mao, L., Li, W., Yang, L., Wang, J., Cheng, S., Wei, Y., Wang, Q., Zhang, W., Hao, F., Ding, Y., Sun, Y. and Jiang, J. 2016. Primary surveys on molecular epidemiology of bovine viral diarrhea virus 1 infecting goats in Jiangsu Province, China. BMC Vet. Res. 12(1), 181. Marques Antunes de Oliveira, A., Stalder, H., Peterhans, E., Sauter, K.S. and Schweizer, M. 2013. Complete genome sequences of both biotypes of a virus pair of bovine viral diarrhea virus subgenotype 1k. Genome Announc. 1(4), e00287-13. Mirshamsy, H., Shafyi, A. and Bahrami, S. 1970. The occurrence of bovine virus diarrhea/mucosal disease in Iran. Arch. Razi Inst. 22, 194–201. Mishra, N., Rajukumar, K., Pateriya, A., Kumar, M., Dubey, P., Behera, S.P., Verma, A., Bhardwaj, P., Kulkarni, D.D., Vijaykrishna, D. and Reddy, Unpublished. 2014. Identification and Molecular Characterization of novel and divergent HoBi-Like pestiviruses from naturally infected cattle in India. Vet. Microbiol. 174, 239–246. Nagai, M., Hayashi, M., Itou, M., Fukutomi, T., Akashi, H., Kida, H. and Sakoda, Y. 2008. Identification of new genetic subtypes of bovine viral diarrhea virus genotype 1 isolated in Japan. Virus Genes 36(1), 135–139. Nagai, M., Ito, T., Sugita, S., Genno, A., Takeuchi, K., Ozawa, T., Sakoda, Y., Nishimori, T., Takamura, K. and Akashi, H. 2001. Genomic and serological diversity of bovine viral diarrhea virus in Japan. Arch. Virol. 146, 685–696. Oguzoglu, T.C., Koc, B.T., Coskun, N., Dogan, F. and Duran-Yelken, S. 2019. Endless variety for bovine virus diarrhea viruses: new members of a novel subgroup into Pestivirus A from Turkey. Trop. Anim. Health Prod. 51(5), 1083–1087. Oguzoglu, T.C., Muz, D., Yilmaz, V., Timurkan, M.O., Alkan, F., Akca, Y. and Burgu, I. 2012. Molecular characteristics of bovine virus diarrhoea virus 1 isolates from Turkey: approaches for an eradication programme. Transbound. Emerg. Dis. 59(4), 303–310. Oguzoglu, T.C., Tan, M.T., Toplu, N., Demir, A.B., Bilge-Dagalp, S., Karaoglu, T., Ozkul, A., Alkan, F., Burgu, I., Haas, L. and Greiser-Wilke, I. 2009. Border disease virus (BDV) infections of small ruminants in Turkey: a new BDV subgroup? Vet. Microbiol. 135, 374–379. Pellerin, C., Van den Hurk, J., Lecomte, J. and Tijssen, P. 1994. Identification of a new group of bovine diarrhea virus strains associated with severe outbreaks and high mortalities. Virology 203, 260–268. Postel, A., Schmeiser, S., Bernau, J., Meindl-Boehmer, A., Pridotkas, G., Dirbakova, Z., Mojzis, M. and Becher, P. 2012. Improved strategy for phylogenetic analysis of classical swine fever virus based on fulllength E2 encoding sequences. Vet. Res. 43(1), 50. Qi, F., Gustad, T., Lewis, T.L. and Berry, E.S. 1993. The nucleotide sequence of the 5—untranslated region of bovine viral diarrhoea virus: its use as a probe in rapid detection of bovine viral diarrhoea viruses and border disease viruses. Mol. Cell. Probes 7, 349–356. Rajput, M.K.S., Abdelsalam, K., Darweesh, M.F., Braun, L.J., Kerkvliet, J., Hoppe, A.D. and Chase, C.C.L. 2017. Both cytopathic and non-cytopathic bovine viral diarrhea virus (BVDV) induced autophagy at a similar rate. Vet. Immunol. Immunopathol. 193–194, 1–9. Ridpath, J.F., Fulton, R.W., Kirkland, P.D. and Neill, J.D. 2010. Prevalence and antigenic differences observed between bovine viral diarrhea virus subgenotypes isolated from cattle in Australia and feedlots in the southwestern United States. J. Vet. Diagn. Invest. 22, 184–191. Saitou, N. and Nei, M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. Sakoda, Y., Ozawa, S., Damrongwatanapokin, S., Sato, M., Ishikawa, K. and Fukusho, A. 1999. Genetic heterogeneity of porcine and ruminant pestiviruses mainly isolated in Japan. Vet. Microbiol. 65, 75–86. Sato, A., Tateishi, K., Shinohara, M., Naoi, Y., Shiokawa, M., Aoki, H., Ohmori, K., Mizutani, T., Shirai, J. and Nagai, M. 2016. complete genome sequencing of bovine viral diarrhea virus 1, subgenotypes 1n and 1o. Genome Announc. 4, e01744-15. Schweizer, M. and Peterhans, E. 1999. Oxidative stress in cells infected with bovine viral diarrhoea virus: a crucial step in the induction of apoptosis. J. Gen. Virol. 80, 1147–1155. Shi, H., Li, H., Zhang, Y., Yang, L., Hu, Y., Wang, Z., Duan, L., Leng, C., Yan, B. and Yao, L. 2020. Genetic diversity of bovine pestiviruses detected in backyard cattle farms between 2014 and 2019 in Henan Province, China. Front. Vet. Sci. 7, 197. Shi, S. and Zhang, X. 1987. Bovine viral diarrhea virus-mucosal disease virus was isolated from frozen semen imported from New Zealand. Chin. J. Vet. Dru. 1, 26–27. Stalder, H.P., Meier, Ph., Pfaffen, G., Wageck-Canal, C., Rüfenacht, J., Schaller, P., Bachofen, C., Marti, S., Vogt, H.R. and Peterhans, E. 2005. Genetic heterogeneity of pestiviruses of ruminants in Switzerland. Prev. Vet. Med. 72, 37–41. Strong, R., Errington, J., Cook, R., Ross-Smith, N., Wakeley, P. and Steinbach, F. 2013. Increased phylogenetic diversity of bovine viral diarrhoea virus type 1 isolates in England and Wales since 2001. Vet. Microbiol. 162(2–4), 315–320. Tajima, M. 2006. The prevalent genotypes of bovine viral diarrhea virus in Japan, Germany and the United States of America. Jpn. J. Vet. Res. 54, 129–34. Tajima, M. 2021. Prevalence of bovine viral diarrhea virus infection in Japan: 2000–2019. Front. Vet. Sci. 8, 667933. Tajima, M., Frey, H.R., Yamato, O., Maede, Y., Moenning, V., Scholz, H. and Greiser-Wilke, I. 2001. Prevalence of genotypes 1 and 2 of bovine viral diarrhea virus in Lower Saxony, Germany. Virus Res. 76, 31–42. Thompson, J.D., Gibson, T.J., Plewniak, F., Jaenmougin, F. and Higgins, D.G. 1997. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4867–4882. Tian, B., Cai, D., Li, W., Bu, Q., Wang, M., Ye, G., Liu, J., Wang, Y., Gou, L., Yi, J. and Zuo, Z. 2021. Identification and genotyping of a new subtype of bovine viral diarrhea virus 1 isolated from cattle with diarrhea. Arch. Virol. 166(4), 1259–1262. Timurkan, M.Ö. and Aydın, H. 2019. Increased genetic diversity of BVDV strains circulating in Eastern Anatolia, Turkey: first detection of BVDV-3 in Turkey. Trop. Anim. Health Prod. 51(7), 1953–1961. Toplu, N., Oguzoglu, T.C. and Albayrak, H. 2012. Dual infection of fetal and neonatal small ruminants with border disease virus and Peste Des Petits Ruminants virus (PPRV): neuronal tropism of PPRV as a novel finding. J. Comp. Pathol. 146, 289–297. Vilček, S., Nettleton, P.F., Paton, D.J. and Belak, S. 1997. Molecular characterization of ovine pestiviruses. J. Gen. Virol. 78, 725–735. Vilček, S., Paton, D.J., Durkovic, B., Strojny, L., Ibata, G., Moussa, A., Loitsch, A., Rossmanith, W., Vega, S., Scicluna, M. and Palfi, V. 2001. Bovine viral diarrhoea virus genotype 1 can be separated into at least eleven genetic groups. Arch. Virol. 146, 99–115. Wang, W., Shi, X., Tong, Q., Wu, Y., Xia, M.Q., Ji, Y., Xue, W. and Wu, H. 2014. A bovine viral diarrhea virus type 1a strain in China: isolation, identification, and experimental infection in calves. Virol. J. 11, 8. Wang, X., Tu, C., Li, H., Xuan, H., Zhu, W., Fei, E. and Yin, Z. 1996. Comparison of the main region within P125 gene of bovine viral diarrhea virus. Chin. J. Vet. Sci. 16(6), 546–553. Weng, X.G., Song, Q.J., Wu, Q., Liu, M.C., Wang, M.L. and Wang, J.F. 2015. Genetic characterization of bovine viral diarrhea virus strains in Beijing, China and innate immune responses of peripheral blood mononuclear cells in persistently infected dairy cattle. J. Vet. Sci. 16(4), 491–500. Xu, X., Zhang, Q., Yu, X., Liang, L., Xiao, C., Xiang, H. and Tu, C. 2006. Sequencing and comparative analysis of a pig bovine viral diarrhea virus genome. Virus Res. 122(1–2), 164–170. Xue, F., Zhu, Y.M., Li, J., Zhu, L.C., Ren, X.G., Feng, J.K., Shi, H.F. and Gao, Y.R. 2010. Genotyping of bovine viral diarrhea viruses from cattle in China between 2005 and 2008. Vet. Microbiol. 143(2–4), 379–383. Yamamoto, T., Kozasa, T., Aoki, H., Sekiguchi, H., Morino, S. and Nakamura, S. 2008. Genomic analyses of bovine viral diarrhea viruses isolated from cattle imported into Japan between 1991 and 2005. Vet. Microbiol. 127(3–4), 386–391. Yesilbag, K., Alpay, G. and Becher, P. 2017. Variability and global distribution of subgenotypes of bovine viral diarrhea virus. Viruses 9(6), 128. Yesilbag, K., Förster, C., Ozyigit, M.O., Alpay, G., Tuncer, P., Thiel, H.-J. and König, M. 2014. Characterisation of bovine viral diarrhoea virus (BVDV) isolates from an outbreak with haemorrhagic enteritis and severe pneumonia. Vet. Microbiol. 169, 42–49. Yitagesu, E., Jackson, W., Kebede, N., Smith, W. and Fentie, T. 2021. Prevalence of bovine abortion, calf mortality, and bovine viral diarrhea virus (BVDV) persistently infected calves among pastoral, peri-urban, and mixed-crop livestock farms in central and Northwest Ethiopia. BMC Vet. Res. 17(1), 87. Zhang, S.Q., Tan, B., Ding, Y., Wang, F., Guo, L., Wen, Y., Cheng, S. and Wu, H. 2014a. Complete genome sequence and pathogenesis of bovine viral diarrhea virus JL-1 isolate from cattle in China. Virol. J. 11, 67. Zhang, S.Q., Tan, B., Guo, L., Wang, F.X., Zhu, H.W., Wen, Y.J. and Cheng, S. 2014b. Genetic diversity of bovine viral diarrhea viruses in commercial bovine serum batches of Chinese origin. Infect. Genet. Evol. 27, 230–233. Zheng, F., Yi, W., Liu, W., Zhu, H., Gong, P. and Pan, Z. 2021. A positively charged surface patch on the Pestivirus NS3 protease module plays an important role in modulating NS3 helicase activity and virus production. Arch. Virol. 166, 1633–1642. Zhong, F., Li, N., Huang, X., Guo, Y., Chen, H., Wang, X., Shi, C. and Zhang, X. 2011. Genetic typing and epidemiologic observation of bovine viral diarrhea virus in Western China. Virus Genes 42(2), 204–207. Zhu, J., Wang, C., Zhang, L., Zhu, T., Li, H., Wang, Y., Xue K., Qi M., Peng, Q., Chen, Y., Hu, C., Chen, X., Chen, J., Chen, H. and Guo, A. 2022. Isolation of BVDV-1a, 1m, and 1v strains from diarrheal calf in China and identification of its genome sequence and cattle virulence. Front. Vet. Sci. 9, 1008107. Zhu, L., Xing, Z., Jia, C., Wang, T., Gai, X., Song, L., Wang, X., Tu, C. and Wang, X. 2014. Complete genomic sequence of bovine viral diarrhea virus strain CC13B. Chin. J. Vet. Sci. 34(7), 1065–1071. Zhu, L., Lu, H., Cao, Y., Gai, X., Guo, C., Liu, Y., Liu, J. and Wang, X. 2016. Molecular Characterization of a Novel Bovine Viral Diarrhea Virus Isolate SD-15. PLoS One 11(10), e0165044. Zuker, M. and Stiegler, P. 1981. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 9, 133–148. Supplementary Material 1. Computing of divergence values based on 5′-UTR secondary structure sequence alignment of BVDV-1 bovine strains EN-6, T4-32, T4-31-2, GA190608, BJ09_26, HY-3, HN1626, HB-03, XH-1, MF-3 and one goat strain JS12/02, reported as genotype 1o or 1v, isolated from China (Mao et al., 2016; Deng et al., 2020; Shi et al., 2020; Tian et al., 2021; Zhu et al., 2022; strain T6-18, reported as genotype 1w isolated from China (Deng et al., 2020), strains TY8723 TR-Erz-Pst8, TRElz-4-2021, TR-Elz-Pst16, IRTV1, and IR-Shiraz-322, reported as genotype 1a, 1l, or 1v, isolated from Turkey and Iran (Oguzoglu et al., 2019; Timurkan and Aydin, 2019; clustered according to PNS method as 1.7.3, 1.19, and 1.21, respectively.
| ||
| How to Cite this Article |
| Pubmed Style Giangaspero M, Zhang S. Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and Turkey.. Open Vet J. 2023; 13(7): 903-931. doi:10.5455/OVJ.2023.v13.i7.12 Web Style Giangaspero M, Zhang S. Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and Turkey.. https://www.openveterinaryjournal.com/?mno=129745 [Access: July 14, 2025]. doi:10.5455/OVJ.2023.v13.i7.12 AMA (American Medical Association) Style Giangaspero M, Zhang S. Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and Turkey.. Open Vet J. 2023; 13(7): 903-931. doi:10.5455/OVJ.2023.v13.i7.12 Vancouver/ICMJE Style Giangaspero M, Zhang S. Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and Turkey.. Open Vet J. (2023), [cited July 14, 2025]; 13(7): 903-931. doi:10.5455/OVJ.2023.v13.i7.12 Harvard Style Giangaspero, M. & Zhang, . S. (2023) Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and Turkey.. Open Vet J, 13 (7), 903-931. doi:10.5455/OVJ.2023.v13.i7.12 Turabian Style Giangaspero, Massimo, and Shuquin Zhang. 2023. Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and Turkey.. Open Veterinary Journal, 13 (7), 903-931. doi:10.5455/OVJ.2023.v13.i7.12 Chicago Style Giangaspero, Massimo, and Shuquin Zhang. "Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and Turkey.." Open Veterinary Journal 13 (2023), 903-931. doi:10.5455/OVJ.2023.v13.i7.12 MLA (The Modern Language Association) Style Giangaspero, Massimo, and Shuquin Zhang. "Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and Turkey.." Open Veterinary Journal 13.7 (2023), 903-931. Print. doi:10.5455/OVJ.2023.v13.i7.12 APA (American Psychological Association) Style Giangaspero, M. & Zhang, . S. (2023) Pestivirus A Bovine viral diarrhea virus type 1 species genotypes circulating in China and Turkey.. Open Veterinary Journal, 13 (7), 903-931. doi:10.5455/OVJ.2023.v13.i7.12 |

